 2022, Vol. 36
2022, Vol. 36



2. 延边大学 药学院, 吉林 延吉 133000
2. Department of Pharmacy, Yanbian University, Yanji 133000, China
δ-羟基-β-酮酸酯骨架结构存在于许多生物活性天然产物和药物中[1-6]. 通常可以利用β-酮酸酯2价阴离子的Aldol反应[7-9]或二烯醇醚、二烯醇硅醚的Mukaiyama Aldol反应获得δ-羟基-β-酮酸酯. 这些方法存在反应条件苛刻、繁琐的保护、去保护过程等问题[10-13]. 关于β-酮酸酯在直接羟醛反应中的报道很有限[14-15]. 2015年, Zhang等[16]报道了1, 8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)催化β-酮酸酯与芳基三氟甲基酮的直接羟醛反应, 选择性发生在β-酮酸酯的γ碳上而得到δ-羟基-β-酮酸酯, 通过拆分的方法获得光学纯产品. 由于3-羟基-3-烷基-2-吲哚酮化合物的生物活性显著, 靛红被作为亲电试剂应用于Aldol反应中制备3-羟基-3-烷基-2-吲哚酮[17-23]. 2013年, Thakur组报道了β-酮酸酯与靛红的Aldol反应, 获得了消旋的δ-羟基-β-酮酸酯[24]. 2020年, Zhang等[25]用金鸡纳碱催化β-酮酸酯与靛红的不对称直接Aldol反应, 制备以3-羟基-2-吲哚酮为骨架结构的手性δ-羟基-β-酮酸酯, 得到了79%~98% ee. 目前仅有1篇关于有机催化β-酮酸酯与靛红的不对称直接Aldol反应的报道, 催化剂种类有限. 我们将一系列Takemoto型(硫)脲催化剂1a-g(图 1)应用于该反应.

|
图 1 手性Takemoto型(硫)脲催化剂1a-g的结构 Fig.1 The structure of chiral Takemoto′s (thio)urea catalysts 1a-g |
催化剂1a-1g购买于上海大赛璐试剂有限公司; 硅胶GF254薄层板及柱色谱分离用粒径0.071~0.050 mm硅胶, 购买于山西诺泰生物科技有限公司; 其他分析纯试剂通过市售渠道购买; 1H NMR和13C NMR光谱通过Bruker Avance-500型核磁共振谱仪(德国Bruker公司)测定; 以氘代DMSO为溶剂, 以未氘代的DMSO为内标(分别为氢谱3.33和碳谱40.45); 高分辨质谱HRMS的测定使用Triple TOF 5600+型质谱仪(美国Sciex公司); 旋光值通过A28579-T-CG APIII型自动旋光仪(美国Rudolph公司)测定; 对映体过量值(ee)的测定使用LC-20A高效液相色谱仪(日本岛津公司)及Daicel ChiralpakIA手性色谱柱(4.6 mm×250 mm, 日本大赛璐公司).
1.2 不对称Aldol反应的一般操作步骤于10 mL带盖试管中加入靛红(0.1 mmol), 乙酰乙酸酯(0.2 mmol), 催化剂1b(0.005 mmol). 然后, 加入1.0 mL甲基叔丁基醚, 充分溶解后, 混合液在0 ℃反应24~48 h, 通过TLC监测反应. 反应完成后, 无需处理, 直接将反应液经硅胶柱层析分离纯化(正己烷: 乙酸乙酯= 9:1), 得到产品3a-3n, 为白色或微黄色固体. 其中产品3b-3e、3j-3k、3n为新化合物, 化合物的1H NMR、13C NMR、HRMS如下:
(S)-4-(1-甲基3-羟基-2-氧吲哚-3-基)-3-氧代丁酸乙酯(3b): 白色固体, mp: 120.1~121.3 ℃.1H NMR(500 MHz, DMSO)δ 7.34 - 7.24(m, 2H), 7.03 - 6.93(m, 2H), 6.13(s, 1H), 4.03(q, J = 7.0 Hz, 2H), 3.56(s, 2H), 3.43(d, J = 17.0 Hz, 1H), 3.14(t, J = 15.0 Hz, 1H), 3.10(s, 3H), 1.14(t, J = 7.0 Hz, 3H); 13C NMR(125 MHz, DMSO)δ 201.1, 177.1, 167.7, 144.9, 131.4, 130.1, 124.2, 122.9, 109.2, 73.1, 61.4, 50.6, 50.2, 26.8, 14.9; HRMS(ESI): calcd for C15H17NO5Na+ [M+Na]+: 314.0999; found: 314.0992; [α]D25= -12.54(c 0.52, CH3OH); HPLC(Chiralpak IA, VC6H14: VC3H8O= 90:10, 1.0 mL/min, λ= 254 nm), tR = 60.8 min(minor), 68.8 min(major).
(S)-4-(5-甲基3-羟基-2-氧吲哚-3-基)-3-氧代丁酸乙酯(3c): 白色固体, mp: 113.8~115.0 ℃. 1H NMR(500 MHz, DMSO)δ 10.14(s, 1H), 7.05(s, 1H), 6.99(d, J = 8.0 Hz, 1H), 6.67(d, J = 8.0 Hz, 1H), 6.02(s, 1H), 4.04(q, J = 7.0 Hz, 2H), 3.67- 3.52(m, 2H), 3.33(d, J = 17.0 Hz, 2H), 3.08(d, J = 17.0 Hz, 1H), 2.23(s, 3H), 1.15(t, J = 7.0 Hz, 3H); 13C NMR(125 MHz, DMSO)δ 201.2, 178.8, 167.8, 140.9, 132.2, 130.9, 130.1, 125.4, 110.1, 73.5, 61.4, 50.5, 50.3, 21.6, 14.9; HRMS(ESI): calcd for C15H17NO5Na+ [M+Na]+: 314.0999; found: 314.0994; [α]D25= -29.67(c 0.62, CH3OH); HPLC(Chiralpak IA, VC6H14: VC3H8O= 90: 10, 1.0 mL/min, λ= 254 nm), tR = 30.7 min(minor), 38.8 min(major).
(S)-4-(5-氟3-羟基-2-氧吲哚-3-基)-3-氧代丁酸乙酯(3d): 微黄色固体, mp: 134.5~136.9 ℃.1H NMR(500 MHz, DMSO)δ 10.27(s, 1H), 7.12(dd, J = 8.0, 2.5 Hz, 1H), 7.01(ddd, J = 9.5, 8.5, 2.5 Hz, 1H), 6.77(dd, J = 8.5, 4.5 Hz, 1H), 6.19(s, 1H), 4.04(q, J = 7.0 Hz, 2H), 3.58(s, 2H), 3.42(d, J = 17.5 Hz, 1H), 3.13(d, J = 17.5 Hz, 1H), 1.15(t, J = 7.0 Hz, 3H); 13C NMR(125 MHz, DMSO)δ 201.2, 178.8, 167.7, 159.7, 157.8, 139.6, 133.8(d, J = 7.7 Hz), 116.2, 116.0(d, J = 23.2 Hz), 112.7, 112.5, 111.1(d, J = 7.8 Hz), 73.6, 61.4, 50.2, 50.1, 14.8; HRMS(ESI): calcd for C14H14FNO5Na+ [M+Na]+: 318.0748; found: 318.0744; [α]D25= -14.0(c 0.49, CH3OH); HPLC Chiralpak IA, VC6H14: VC3H8O= 90: 10, 1.0 mL/min, λ= 254 nm), tR = 32.5 min(minor), 42.3 min(major).
(S)-4-(5-氯3-羟基-2-氧吲哚-3-基)-3-氧代丁酸乙酯(3e): 微黄色固体, mp: 140.2~141.6 ℃.1H NMR(500 MHz, DMSO)δ 10.38(s, 1H), 7.29(d, J = 2.0 Hz, 1H), 7.23(dd, J = 8.0, 2.0 Hz, 1H), 6.79(d, J = 8.0 Hz, 1H), 6.18(s, 1H), 4.04(q, J = 7.0 Hz, 2H), 3.58(d, J = 2.0 Hz, 2H), 3.46(d, J = 17.5 Hz, 1H), 3.15(d, J = 17.5 Hz, 1H), 1.14(t, J = 7.0 Hz, 3H); 13C NMR(125 MHz, DMSO)δ 201.3, 178.5, 167.7, 142.4, 134.3, 129.7, 126.2, 125.0, 111.8, 73.4, 61.4, 50.2 50.1, 14.9; HRMS(ESI): calcd for C14H14ClNO5Na+ [M+Na]+: 334.0453; found: 334.0459; [α]D25= -9.98(c 0.50, CH3OH); HPLC(Chiralpak IA, VC6H14: VC3H8O= 90: 10, 1.0 mL/min, λ= 254 nm), tR = 33.2 min(minor), 44.2 min(major).
(S)-4-(5-甲基3-羟基-2-氧吲哚-3-基)-3-氧代丁酸甲酯(3j): 白色固体, mp: 105.3~106.7 ℃.1H NMR(500 MHz, DMSO)δ 10.14(s, 1H), 7.05(s, 1H), 7.04-6.94(m, 1H), 6.67(d, J = 8.0 Hz, 1H), 6.02(s, 1H), 3.61(s, 2H), 3.58(s, 3H), 3.32(d, J = 17.5 Hz, 2H), 3.08(d, J = 17.5 Hz, 1H), 2.23(s, 3H); 13C NMR(125 MHz, DMSO)δ 201.1, 178.8, 168.3, 140.9, 132.2, 130.9, 130.2, 125.4, 110.1, 73.5, 52.7, 50.5, 50.1, 21.6; HRMS(ESI): calcd for C14H15NO5Na+ [M+Na]+: 300.0842; found: 300.0846; [α]D25= -20.27(c 0.68, CH3OH); HPLC(Chiralpak IA, VC6H14: VC3H8O= 90: 10, 1.0 mL/min, λ= 254 nm), tR = 36.0 min(minor), 45.0 min(major).
(S)-4-(5-氯3-羟基-2-氧吲哚-3-基)-3-氧代丁酸甲酯(3k): 微黄色固体, mp: 130.7~132.0 ℃.1H NMR(500 MHz, DMSO)δ 10.38(s, 1H), 7.30(d, J = 2.0 Hz, 1H), 7.23(dd, J = 8.0, 2.0 Hz, 1H), 6.79(d, J = 8.0 Hz, 1H), 6.19(s, 1H), 3.61(s, 2H), 3.58(s, 3H), 3.46(d, J = 17.5 Hz, 1H), 3.15(d, J = 17.5 Hz, 1H); 13C NMR(125 MHz, DMSO)δ 201.3, 178.5, 168.2, 142.4, 134.2, 129.7, 126.2, 125.0, 111.8, 73.4, 52.7, 50.1, 49.9; HRMS(ESI): calcd for C13H12ClNO5Na+ [M+Na]+: 320.0296; found: 320.0299; [α]D25= -38.33(c 0.56, CH3OH); HPLC(Chiralpak IA, VC6H14: VC3H8O= 90: 10, 1.0 mL/min, λ= 254 nm), tR = 38.7 min(minor), 52.2 min(major).
(S)-4-(5-氯3-羟基-2-氧吲哚-3-基)-3-氧代丁酸丁酯(3n): 微黄色固体, mp: 124.7~126.0 ℃. 1H NMR(500 MHz, DMSO)δ 10.26(s, 1H), 7.22(d, J = 7.5 Hz, 1H), 7.17(dd, J = 7.5, 1.0 Hz, 1H), 6.93- 6.88(m, 1H), 6.78(d, J = 7.5 Hz, 1H), 6.08(s, 1H), 3.99(t, J = 6.5 Hz, 2H), 3.59(s, 2H), 3.38(s, 1H), 3.07(d, J = 17.0 Hz, 1H), 1.54- 1.46(m, 2H), 1.33-1.25(m, 2H), 0.86(t, J = 7.5 Hz, 3H); 13C NMR(125 MHz, DMSO)δ 201.2, 178.8, 167.9, 143.4, 132.1, 130.0, 124.7, 122.2, 110.4, 73.4, 65.1, 50.5, 50.3, 31.0, 19.4, 14.4; HRMS(ESI): calcd for C16H19NO5Na+ [M+Na]+: 328.1155; found: 328.1161; [α]D25= -0.32(c 0.51, CH3OH); HPLC(Chiralpak IA, VC6H14: VC3H8O= 90: 10, 1.0 mL/min, λ= 254 nm), tR = 26.6 min(minor), 36.0 min(major).
2 结果与讨论 2.1 Takemoto型(硫)脲1a-g催化不对称Aldol反应分别将10%(摩尔分数)(硫)脲衍生物1a-g用于有机催化靛红与乙酰乙酸乙酯的不对称Aldol反应, 根据文献[25]的最佳反应条件, 采用四氢呋喃为溶剂, 室温反应24~48 h(TLC监测反应),结果见表 1.
| 表 1 (硫)脲1a-g催化靛红与乙酰乙酸乙酯的不对称Aldol反应的结果a Table 1 Asymmetric Aldol reaction of isatin with ethyl acetoacetate catalyzed by (thio)ureas 1a-ga |
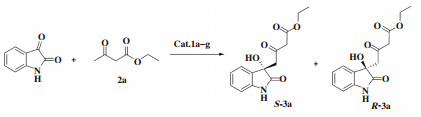
表 1结果显示: 除了催化剂1g, 其他6种(硫)脲催化剂在四氢呋喃中均能顺利催化靛红和乙酰乙酸乙酯的不对称Aldol反应获得δ-羟基-β-酮酸乙酯产物, 得到了50%~64%的立体选择性和77%~83%的产率. 其中, 脲衍生物催化剂1b表现出最优的不对称诱导作用, 其得到主产物的构型通过测定旋光值并与文献值对比[25], 确定为S构型.
2.2 反应条件的考察将上述筛选出的最佳催化剂1b用于不同溶剂、温度、催化剂用量等条件下靛红和乙酰乙酸乙酯的不对称Aldol反应中, 考察反应条件对立体选择性的影响, 结果见表 2.
| 表 2 1b催化靛红和乙酰乙酸乙酯不对称Aldol反应的条件筛选a Table 2 Screening of reaction condition for the asymmetric Aldol reaction catalyzed by 1ba |
我们考察了8种溶剂对反应的立体选择性的影响, 可以看出在醚类溶剂中该反应表现出良好的对映选择性(entries 1, 4-6), 明显优于其他种类的溶剂. 其中, 甲基叔丁基醚(MTBE)为最佳溶剂(66% ee, entry5).温度对反应的立体选择性有影响, 通常低温有利于降低反应速率, 提高反应的选择性, 但是, 往往也会影响反应的产率. 我们首先将温度由室温降至0 ℃, 导致反应的对映选择性提高了9%, 而产率略有下降(entry 9 vs entry 5); 进而, 继续降温至-10、-20和-40 ℃, 结果表明产率和ee值均有所降低(entry 10-12 vs entry 9). 在筛选出的最适合的溶剂和温度条件下, 继续考察催化剂用量对反应的立体选择性的影响, 当催化剂摩尔分数由10%降至5%, 产品的对映体过量值升高12%(entry 13 vs entry 9). 使用更少量的催化剂得到更好的立体选择性是筛选催化剂的重要指标, 能够降低反应的成本, 提高其实用价值. 但是, 继续降低催化剂摩尔分数至2.5%, 产品的ee值和产率均明显降低(entry 14 vs entry 13). 此外, 将催化剂的摩尔分数由10%增至20%, 产率稍有提高, 但是反应的立体选择性有所下降(entry15 vs entry 13).通常反应溶液稀释能够降低反应速率, 提高反应的选择性, 因此, 我们将溶剂量加倍(2 mL), 结果表明反应的产率和立体选择性均明显降低(entry 16 vs entry 13). 最后, 我们考察了不对称反应中常用的0.4 nm分子筛对该反应立体选择性的影响, 结果导致催化剂的不对称诱导作用明显降低(entry 17vs entry 13). 综上所述, 筛选出最佳催化剂体系为: 5%(摩尔分数)催化剂1b, 甲基叔丁基醚(1 mL), 0 ℃反应.
2.3 普适性的考察为了考察上述最佳催化剂体系的普适性, 扩展了该反应中底物的范围, 将8种不同取代靛红和4种不同的乙酰乙酸酯应用于不对称Aldol反应, 结果见表 3.
| 表 3 不同乙酰乙酸酯与不同取代靛红的不对称Aldol反应a Table 3 Generility of the enantioselective Aldol reaction of isatins with acetoacetates a |
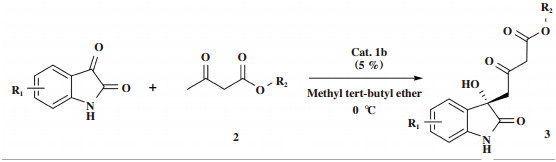
结果表明: 催化剂1b能够催化不同取代靛红与乙酰乙酸甲(乙)酯的不对称Aldol反应, 以76%~87%的产率得到相应的产品. 其中, 靛红与乙酰乙酸乙酯的反应得到了最高的ee值(87% ee, entry1). 靛红的取代基位置对反应的立体选择性有影响. 5位取代靛红的反应表现出较高的对映选择性(entries 3-6). 4位和7位取代靛红的反应仅得到了51%和56%的ee值.当靛红或相同取代靛红分别与乙酰乙酸乙酯及甲酯反应时, 乙酯反应的立体选择性明显优于甲酯的反应结果(entries 1, 3, 5 vs entries 9-11).由于不同乙酰乙酸酯底物对反应立体选择性有影响, 我们又将乙酰乙酸异丙酯和丁酯作为底物分别与靛红进行反应, 两种酯均得到了82% ee(entries 12, 14), 结果优于甲酯(71% ee), 但是低于乙酯(87% ee).可能是电性效应和空间效应综合作用的结果.因此, 靛红结构中取代基的种类和位置以及乙酰乙酸酯的种类对反应的立体选择性均有一定的影响.
3 结论综上所述, 将Takemoto型(硫)脲催化剂用于有机催化靛红与乙酰乙酸乙酯的不对称直接Aldol反应.筛选出最佳的催化条件, 并应用于多种底物的Aldol反应, 以最高达87%的对映选择性获得了手性δ-(2-羟基吲哚-3基)-δ-羟基-β-酮酸酯.扩展了催化剂类型和底物范围. 影响反应立体选择性的机制还有待于进一步研究.
| [1] |
Zacharia J T, Tanaka T, Hayashi M. Facile and highly enantioselective synthesis of (+)- and (-)-fluvastatin and their analogues[J]. J Org Chem, 2010, 75(22): 7514–7518.
DOI:10.1021/jo101542y |
| [2] |
a. Clarke P A, Santos S, Mistry N, et al. The asymmetric maitland-japp reaction and its application to the construction of the C1-C19 Bis-pyran unit of phor-boxazole B[J]. Org Lett, 2011, 13(4): 624-627. b. Jiang He-yan(蒋和雁), Cheng Hong-mei(成洪梅), Chen Shi-jia(陈诗佳), et al. Efficient asymmetric hydr-ogenation of α-ketoesters catalyzed by cinchona alkaloid and ionic liquid Co-stabilized Pt nanoparticles(离子液中金鸡纳碱稳定的Pt纳米粒子催化α-酮酸酯不对称加氢)[J]. J Mol Catal(China)(分子催化), 2020, 34(3): 193-200. c. An Shi-yun(安士云), Wei Zhao(魏钊), Zhang Jin-long(张金龙), et al. Recent progress in asymmetric cat-alytic friedel-crafts reaction of pyrroles (基于吡咯骨架的不对称傅克反应研究进展)[J]. J Mol Catal(China)(分子催化), 2020, 34(3): 261-271. |
| [3] |
Clarke P A, Nasir N M, Sellars P B, et al. Synthesis of 2, 6-trans- and 3, 3, 6-trisubstituted tetrahydropyran-4-ones from Maitland–Japp derived 2H-dihydropyran-4-ones: A total synthesis of diospongin B[J]. Org Biomol Chem, 2016, 14(28): 6840–6852.
DOI:10.1039/C6OB01182A |
| [4] |
Puppala M, Narayanapillai S C, Leitzman P, et al. Pilot in vivo structure-activity relationship of dihydromethysticin in blocking 4-(methylnitrosamino)-1-(3-pyridyl) -1-butanone- Induced O-methylguanine and lung tumor in A/J Mice[J]. J Med Chem, 2017, 60(18): 7935–7940.
DOI:10.1021/acs.jmedchem.7b00921 |
| [5] |
Cheng H, Zhang Z H, Yao H L, et al. Unified asymm-etric total syntheses of (-)-alotaketals A-D and (-)-Phorbaketal A[J]. Angew Chem, Int Ed, 2017, 56(31): 9096–9100.
DOI:10.1002/anie.201704628 |
| [6] |
Bjerketorp J, Levenfors J J, Sahlberg C, et al. Antiba-cterial 3, 6-disubstituted 4-hydroxy-5, 6- dihydro-2H-pyran-2-ones from Serratia plymuthica MF371-2[J]. J Nat Prod, 2017, 80(11): 2997–3002.
DOI:10.1021/acs.jnatprod.7b00565 |
| [7] |
Kang T, Jo D, Han S. Six-step total synthesis of azaspirene[J]. J Org Chem, 2017, 82(18): 9335–9341.
DOI:10.1021/acs.joc.7b01224 |
| [8] |
Jo D, Han S. Biomimetic total synthesis of (±)-ber-keleyamide D[J]. Org Chem Front, 2017, 4(4): 506–509.
DOI:10.1039/C6QO00812G |
| [9] |
Smrcček J, Pohl R, Jahn U. Total syntheses of All tri-oxygenated 16-phytoprostane classes via a common prec-ursor constructed by oxidative cyclization and alkyl-alkyl coupling reactions as the key steps[J]. Org Biomol Chem, 2017, 15(44): 9408–9414.
DOI:10.1039/C7OB02505J |
| [10] |
Casiraghi G, Battistini L, Curti C, et al. The vinylogous aldol and related addition reactions: Ten years of progress[J]. Chem Rev, 2011, 111(5): 3076–3154.
|
| [11] |
Pansare S V, Paul E K. The organocatalytic vinylogous aldol reaction: Recent advances[J]. Chem Eur J, 2011, 17(32): 8770–8779.
DOI:10.1002/chem.201101269 |
| [12] |
Clarke P A, Ermanis K. Synthesis of the C20-C32 tetrahydropyran core of the phorboxazoles and the C22 epimer via a stereodivergent michael reaction[J]. Org Lett, 2012, 14(21): 5550–5553.
DOI:10.1021/ol3026523 |
| [13] |
Mikame Y, Yoshida K, Hashizume D, et al. Synthesis of all stereoisomers of RK460 and evaluation of their activity and selectivity as abscisic acid receptor antagonists[J]. Chem Eur J, 2019, 25(14): 3496–3500.
DOI:10.1002/chem.201806056 |
| [14] |
Benetti S, Romagnoli R, De Risi C, et al. Mastering beta-Keto Esters[J]. Chem Rev, 1995, 95(4): 1065–1114.
DOI:10.1021/cr00036a007 |
| [15] |
Govender T, Arvidsson P I, Maguire G E M, et al. Enan-tioselective organocatalyzed transformations of β-keto-esters[J]. Chem Rev, 2016, 116(16): 9375–9437.
DOI:10.1021/acs.chemrev.6b00156 |
| [16] |
Zhang D X, Tanaka F. Aldol reactions of ketone donors with aryl trifluoromethyl ketone acceptors catalyzed by 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) for concise access to aryl- and trifluoromethyl- substituted tertiary alcohols[J]. Adv Synth Catal, 2015, 357(16/17): 3458–3462.
|
| [17] |
Raviraj A, Manohar V. Synthesis of spiro[indolo-1, 5-benzodiazepines] from 3-acetyl coumarins for use as possible antianxiety agents[J]. J Chem Sci, 2004, 116(5): 265–270.
DOI:10.1007/BF02708277 |
| [18] |
Guo Q S, Bhanushali M, Zhao C G. Quinidine thiourea-catalyzed aldol reaction of unactivated ketones: Highly enantioselective synthesis of 3-alkyl-3-hydro- xyin-dolin-2-ones.[J]. Angew Chem, Int Ed, 2010, 49(49): 9460–9464.
DOI:10.1002/anie.201004161 |
| [19] |
Abbaraju S. Asymmetric aldol reaction of 3-acety-2H-chromen-2-ones and isatins catalyzed by a bifunctional quinidine urea catalyst[J]. Adv Synth Catal, 2014, 356(1): 237–241.
DOI:10.1002/adsc.201300623 |
| [20] |
Allu S, Molleti N, Panem R, et al. Enantioselective organ-ocatalytic aldol reaction of unactivated ketones with isatins[J]. Tetrahedron Lett, 2011, 52(32): 4080–4083.
DOI:10.1016/j.tetlet.2011.05.013 |
| [21] |
Lu H F, Bai J T, Xu J Y, et al. Chiral 2-aminopyrimidin-4(1H)-one derivative as the novel bifunctional organocatalyst: Enantioselective aldol reaction of isatins with ketones[J]. Tetrahedron, 2015, 71(18): 2610–2615.
DOI:10.1016/j.tet.2015.03.053 |
| [22] |
Kimura J, Reddy U V S, Kohari Y, et al. Simple primary amino amide organocatalyst for enantioselective aldol reactions of isatins with ketones[J]. Eur J Org Chem, 2016, 2016(22): 3748–3756.
DOI:10.1002/ejoc.201600414 |
| [23] |
Wang L M, Zhao M J, Chen Z, et al. Urea derivative cata-lyzed enantioselective aldol reaction of isatins with ketones[J]. Chirality, 2018, 30(8): 1005–1011.
DOI:10.1002/chir.22977 |
| [24] |
Thakur P B, Sirisha K, Sarma A V S, et al. Highly regios-elective and metal-free γ-addition of β-ketoesters to isatins, catalyzed by DABCO: Direct access to novel class of diversely functionalized 3-hydroxy-2-oxindole scaffolds[J]. Tetrahedron, 2013, 69(31): 6415–6423.
DOI:10.1016/j.tet.2013.05.101 |
| [25] |
Zhang D X, Chen Y, Cai H, et al. Direct catalytic asymmetric synthesis of oxindole-derived δ-Hydroxy-β-ketoesters by aldol reactions[J]. Org Lett, 2020, 22(1): 6–10.
DOI:10.1021/acs.orglett.9b03527 |