 2022, Vol. 36
2022, Vol. 36



线性α-烯烃(Linear Alpha Olefins, LAOs)是分子结构中含有唯一末端双键的C4及C4以上的直链碳氢化合物. LAOs是一类用途广泛的有机化工原料和中间体. 在工业生产中, 以石蜡为原料的裂解工艺可以得到C5~C20+的直链α-烯烃, 或者从费托法煤制油工艺中通过萃取分离得到碳数连续分布的高品质α-烯烃. 生产LAOs的方法以原料的不同可划分为两类: (1)以石蜡、煤等为原料, 生产的LAOs中既有偶数碳的α-烯烃也有奇数碳的α-烯烃; (2)以乙烯为原料, 生产碳原子数为偶数的α-烯烃. 采用乙烯为原料生产LAOs的工艺也分为两类: (1)非选择性齐聚, 获得的产品是C4~C30+的全分布α-烯烃(雪佛龙菲利普斯化工公司的Gulf工艺(Ziegler一步法)、乙基公司的Ethyl工艺(Ziegler两步法)等); (2)选择性齐聚, 获得的产品是具有特定碳数的线性α-烯烃(雪佛龙菲利普斯化工公司的铬系乙烯三聚工艺、大庆石化公司1-己烯生产工艺等)[1]. LAOs下游领域包括聚乙烯共聚单体(C4~C8)、合成润滑油(C8~C12)、表面活性剂(C12~C18)、增塑剂用醇(C6~C10)等.
当前工业生产中LAOs装置以乙烯非选择性齐聚工艺为主. 下游市场对α-烯烃有着不同的需求, 其中C4~C8组分作为线性低密度聚乙烯和高密度聚乙烯的共聚单体的消费量占全球线性α-烯烃消费量的一半以上. 其中, 1-己烯和1-辛烯作为共聚单体可以赋予产品更优异的性能和更高的附加值. 近年来, 市场对高性能聚乙烯的需求, 进一步拉大了1-己烯和1-辛烯与其他α-烯烃在下游市场的需求差异. 因此, 生产全馏分α-烯烃的传统非选择性齐聚工艺获得的产品分布无法满足市场的需求, 这种市场驱动力使得乙烯选择性三聚/四聚成为乙烯齐聚领域的研究热点.
针对乙烯选择性三聚/四聚的研究, 已有论文从不同的角度综述了乙烯选择性三聚/四聚的进展[2-11]. 在现有金属催化体系中, 铬系催化体系是研究最为广泛的乙烯选择性三聚/四聚催化体系. 我们从配体、铬源、助催化剂、反应条件等方面综述了典型的铬系乙烯选择性三聚/四聚催化体系, 并简述了乙烯选择性三聚/四聚的反应机理.
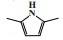
1 铬系乙烯三聚催化体系 1.1 菲利普斯催化体系菲利普斯催化体系是首次实现1-己烯选择性大于90%的乙烯三聚催化体系[12]. 20世纪80年代, 菲利普斯石油公司(Phillips Petroleum Company)在乙烯聚合催化剂铬环戊二烯基配合物的启发下, 研究了铬吡咯化合物的催化性能. 其中, 2, 5-二甲基吡咯(DMP)因其较高的空气、光、温度稳定性以及优异的催化性能成为优选配体. 以1∶3.3∶7.8∶10.8的摩尔比在甲苯溶剂中混合2-乙基己酸铬(Cr(EH)3)、2, 5-二甲基吡咯、二乙基氯化铝和三乙基铝制备的催化剂, 在115 ℃和10.0 MPa的反应条件下, 催化活性为155 246 g/(gCr·h), 1-己烯的选择性达到93.1%, 主要副产物是直链癸烯和支链癸烯(图 1)[13]. 2003年, 雪佛龙菲利普斯化工公司(Chevron Phillips Chemical Company)在卡塔尔建成了世界上首个乙烯选择性三聚工业化装置.

|
图 1 菲利普斯乙烯选择性三聚催化体系 Fig.1 Phillips catalytic system for ethylene trimerization |
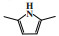
随后, 三菱化学公司(Mitsubishi Chemical Corporation)对该体系进行改进[14]. 将2-乙基己酸铬、2, 5-二甲基吡咯、六氯乙烷和三乙基铝以1∶6∶4∶40的摩尔比混合, 并控制反应器内部的1-己烯与乙烯的摩尔比低于0.5. 在105 ℃和4.9 MPa的反应条件下, 该体系的反应活性为5 391 667 g/(gCr·h), 1-己烯的选择性为96.3%(图 2). 三菱化学公司改进后的催化体系不仅提高了1-己烯的选择性, 而且反应活性是菲利普斯催化体系的30多倍.

|
图 2 经三菱改进的乙烯选择性三聚催化体系 Fig.2 Mitsubishi-modified catalytic system for ethylene trimerization |
除了三菱化学公司, 其他公司以及研究团队也探讨了菲利普斯催化体系或者其变体[15-16]. 在均相Cr(EH)3/DMP/TEA/氯化合物催化体系中, 随着氯化合物中氯取代数量的增加, 观察到1-己烯选择性和乙烯转化率提高. 而含有两个氯的氯化合物大大提高了乙烯三聚的催化活性[15]. 对比研究表明, 氯化物的促进效果明显优于氟化物或溴化物, 促进效果最好的卤化物是多氯化合物[17]. DMP/Cr(Ⅲ)/烷基铝/四氯乙烷催化体系催化乙烯三聚反应的结果表明, 三乙基铝(TEA)、三甲基铝(TMA)、三正己基铝(TNHA)和三异丁基铝(TIBA)都是乙烯三聚的有效助催化剂. 在80 ℃和2.0 MPa的反应条件下, DMP/Cr(Ⅲ)/TEA/TCE催化体系的1-己烯选择性最高为92.4%, 活性为26 540 g/(gCr·h)(原文为1 380 000 g/(molCr·h))[18]. 添加H2对2-乙基己酸铬/2, 5-二甲基吡咯/三乙基铝/四氯乙烷催化体系1-己烯选择性的影响并不明显, 但减少了聚合物的形成并提高了活性[19]. 为了理解乙烯三聚的反应机理, 科研人员利用密度泛函理论(DFT)研究了基本组分(即配体、助催化剂和卤代烃化合物)对菲利普斯催化体系催化乙烯三聚反应的影响[20-21].
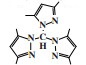
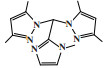
1.2 Cr-NNN体系东曹公司(Tosoh Corporation)的研究人员在四氢呋喃(THF)中将三(3, 5-二甲基-1-吡唑)甲烷配体与CrCl3(THF)3反应制备Cr-NNN配合物, 在80 ℃和4.0 MPa乙烯压力下, 经甲基铝氧烷(MAO)活化后, 1-己烯的选择性可达99.1%, 反应活性为40 100 g/(gCr·h)(图 3)[22]. 随后, Li等[23]合成的Cr-NNN(1, 3, 5-三氮杂环己烷)配合物经MAO活化后, 在50 ℃和0.8 MPa压力下可催化乙烯齐聚得到93.1%的总1-己烯和1-辛烯选择性.

|
图 3 Cr-NNN乙烯选择性三聚催化体系 Fig.3 Cr-NNN catalytic system for ethylene trimerization |
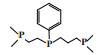
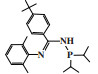
阿莫科公司(Amoco Corporation)使用(2-二甲基膦乙基)(3-二甲基膦丙基)苯基膦配体与CrCl3合成的Cr-PPP配合物经丁基铝氧烷活化后, 在60 ℃和3.5 MPa乙烯压力的反应条件下, 催化活性达到了26 225 g/(gCr·h)(原文为48 700 mol乙烯/(molCr·h)), 1-己烯的选择性为97.3%(图 4)[24].

|
图 4 Cr-PPP乙烯选择性三聚催化体系 Fig.4 Cr-PPP catalytic system for ethylene trimerization |
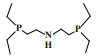
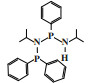
英国石油公司(BP)开发了一种双磷PNP配体的铬系乙烯选择性三聚催化剂. 以CrCl3(THF)3为铬源、Ar2PN(Me)PAr2(Ar =(o-OMe)C6H4)为配体、MAO为活化剂、甲苯为溶剂, 在80 ℃和2.0 MPa压力下催化乙烯三聚得到89.9%的1-己烯, 催化活性为1 033 200 g/(gCr·h)(图 5)[25].

|
图 5 Cr-PNP乙烯选择性三聚催化体系 Fig.5 Cr-PNP catalytic system for ethylene trimerization |
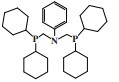
萨索尔公司(Sasol Technology)对BP的PNP配体进行改性, 开发了一种R2P(CH2)2N(H)(CH2)2PR2型三齿配体的铬系乙烯三聚催化剂. 以CrCl3(THF)3为铬源、二乙基膦基乙基胺为配体、MAO为活化剂、甲苯为溶剂, 在100 ℃和4.0 MPa压力的反应条件下, 1-己烯选择性为97.2%, 催化活性为9331 g/(gCr·h)(图 6)[26]. 2009年, Klemps等[27]提出了一种含二齿双膦甲基胺(R’N(CH2PR2)2)的铬配合物. Cr-PCNCP配合物经MAO活化后, 在70 ℃、3.0 MPa乙烯压力下催化乙烯三聚反应, 获得了97.0%的1-己烯, 反应活性为9783 g/(gCr·h)(图 7).

|
图 6 Cr-PC2NC2P乙烯选择性三聚催化体系 Fig.6 Cr-PC2NC2P catalytic system for ethylene trimerization |

|
图 7 Cr-PCNCP乙烯选择性三聚催化体系 Fig.7 Cr-PCNCP catalytic system for ethylene trimerization |
Cr-PNP催化体系出现后, 许多研究人员探索了反应条件、助催化剂、配体主链、配体上的N-取代基或芳基取代基等对Cr-PNP体系催化乙烯三聚反应的影响[28-40], 并通过实验和理论计算(DFT)方法研究了Cr-PNP体系的催化反应机理[41-49]. Blann等[33]的实验表明将苯环上的邻位甲氧基取代基改为甲基、乙基、异丙基后, 该体系仍然是乙烯三聚催化剂, 1-己烯的选择性均超过90%. Alam等[37-38]向PNP配体的N-取代基上引入环戊烷、向一个P-取代基上引入两个N(R2)(R=Et或者iPr)基团也可得到1-己烯选择性超过97%的Cr-PNP催化剂. Liu等[50]使用密度泛函理论方法研究了H2对Cr-PNP体系催化乙烯聚合反应选择性的影响, 发现H2与铬中心的缔合/解离作用可能是该系统高活性和低1-辛烯选择性的关键因素. 目前, 超支化铬系催化剂也得到了众多研究者的关注[51-53].
1.5 Cr-SNS体系McGuinness等[54]利用SNS配体与CrCl3(THF)3合成了一种新型乙烯三聚催化剂. 在90 ℃和3.0 MPa反应条件下, 经MAO活化后进行乙烯三聚反应, 得到98.1%的1-己烯, 催化活性为160 840 g/(gCr·h)(图 8). Cr-SNS催化体系需要加入的MAO量较少, 且SNS配体的合成相对简单, 是一种良好的乙烯三聚工业化候选催化体系.

|
图 8 Cr-SNS乙烯选择性三聚催化体系 Fig.8 Cr-SNS catalytic system for ethylene trimerization |
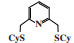
Temple等[55]对Cr-SNS体系进行了进一步的研究, 发现使用2, 6-(CySCH2)2吡啶配体与CrCl3(THF)3合成的新型Cr-SNS配合物经MAO活化后, 在50 ℃和3.5 MPa反应条件下, 1-己烯的选择性高达99.8%, 催化剂活性为3883 g/(gCr·h)(图 9). 与萨索尔公司的Cr-SNS[54]相比, 两个体系1-己烯的选择性相近. 因此作者认为N-H官能团并不是Cr-SNS体系高1-己烯选择性的主要影响因素.

|
图 9 改性的Cr-SNS乙烯选择性三聚催化体系 Fig.9 Modified Cr-SNS catalytic system for ethylene trimerization |
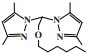
Zhang等[56]使用Pz2CHCH2OR(Pz =吡唑-1-基, R =己基)配体与CrCl3(THF)3合成了一种Cr-NON配合物, 经MAO活化后, 在80 ℃和2.5 MPa反应条件下进行乙烯三聚反应. 该体系反应活性为16 200 g/(gCr·h), 1-己烯的选择性为98.5%(图 10). 在相同的反应条件下, 使用Pz2CHCH2SR(Pz =吡唑-1-基, R =甲基)配体也可得到95.5%的选择性, 但活性仅有2800 g/(gCr·h).

|
图 10 Cr-NON乙烯选择性三聚催化体系 Fig.10 Cr-NON catalytic system for ethylene trimerization |
Zhang等[57]继续探究了配体中第3供体基团(吡啶基、吡唑基、咪唑基以及胺基)对乙烯三聚反应的影响, 所有合成的Cr-NNN体系在MAO活化后都表现出乙烯三聚催化活性. 其中含有咪唑基的配合物, 在80 ℃和3.0 MPa的反应条件下表现出了53 000 g/(gCr·h)的最高活性和97.6% 的1-己烯选择性(图 11).

|
图 11 Cr-NNN乙烯选择性三聚催化体系 Fig.11 Cr-NNN catalytic system for ethylene trimerization |
Albahily等[58]使用NPN配体与CrCl2(THF)2或CrCl3(THF)3合成了一种用于乙烯三聚的Cr-NPN催化体系. 该体系以AliBu3为助催化剂、在50 ℃和3.5 MPa反应条件下获得了 > 99.9%的1-己烯选择性, 反应活性为7693 g/(gCr·h)(原文为400 g/(mmolCr·h), 图 12).

|
图 12 Cr-NPN乙烯选择性三聚催化体系 Fig.12 Cr-NPN catalytic system for ethylene trimerization |
Sydora等[59]使用N-膦基脒配体和CrCl3(THF)3合成的一种Cr-PNCN配合物经MMAO活化后, 在70 ℃、0.2 MPa氢气和6.0 MPa乙烯压力的反应条件下获得了93.6%的1-己烯, 反应活性为1 052 002 g/(gCr·h)(原文为54 700 kg/(molCr·h), 图 13). 随后, Sydora等使用实验方法或者DFT方法继续研究P-取代基、C-取代基、配位模式等对Cr-PNCN催化剂体系的1-己烯和1-辛烯选择性的影响[60-62].

|
图 13 Cr-PNCN乙烯选择性三聚催化体系 Fig.13 Cr-PNCN catalytic system for ethylene trimerization |
德国林德公司(Linde AG)和沙特基础工业公司(SABIC)的研究人员[63]使用Ph2PN(iPr)P(Ph)N(iPr)H配体与CrCl3(THF)3合成的Cr-PNPNH配合物经Et3Al活化后, 在65 ℃和3.0 MPa乙烯压力的反应条件下获得了90.1%的1-己烯, 反应活性为5200 g/(gCr·h)(图 14). 这一体系具有选择性高、催化剂稳定性高、使用低成本的Et3Al以及聚乙烯含量低等优点, 也是一种良好的乙烯三聚工业化候选催化体系.

|
图 14 Cr-PNPNH乙烯选择性三聚催化体系 Fig.14 Cr-PNPNH catalytic system for ethylene trimerization |
表 1按1-己烯的选择性从高到低依次列出了典型的高选择性乙烯三聚催化体系, 其中1-己烯选择性最高可达99.9%. 除已工业化的菲利普斯催化体系之外, Cr-PNP催化体系是研究最为广泛, 受关注程度最高的乙烯三聚催化体系. 高选择性乙烯三聚催化体系最常用的铬源和溶剂分别是CrCl3(THF)3和甲苯. 乙烯三聚反应通常以MAO为助催化剂, 但关于助催化剂作用机理方面的研究相对薄弱. 高选择性乙烯三聚催化剂的反应温度普遍在50 ℃以上, 反应压力范围为2.0 ~ 10.0 MPa的中等压力.
| 表 1 典型的铬系乙烯三聚催化体系 Table 1 Chromium-based catalysts for ethylene trimerization |
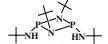

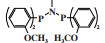
2004年, 萨索尔公司的Bollmann等报道了第一个高选择性乙烯四聚催化体系, 该体系通过调整Cr-PNP配体中N-取代基以及P-取代基实现了较高的1-辛烯选择性[64]. 在众多候选配体中, N-取代基为异丙基、P-取代基为苯基的候选配体与Cr(acac)3合成的Cr-PNP配合物经MAO活化后, 在45 ℃和4.5 MPa反应条件下获得了67.5%的1-辛烯选择性, 催化活性为272 400 g/(gCr·h)(图 15). Cr-NiPr(PPh2)2催化体系经乙基铝氧烷/三甲基铝(EAO/TMA)活化后, 在45 ℃和4.5 MPa反应条件下获得了70.5%的1-辛烯, 但催化活性仅有之前的1/10. 该体系的主要副产物是甲基环戊烷和亚甲基环戊烷.

|
图 15 Cr-PNP乙烯选择性四聚催化体系 Fig.15 Cr-PNP catalytic system for ethylene tetramerization |
随后, 众多研究人员探索了N-取代基、苯环取代基等对Cr-PNP乙烯选择性四聚体系活性和选择性的影响[33-34, 65-74]. Blann等[65]研究了N-取代基对体系选择性的影响, 向N上引入大体积的烷基取代基后体系仍然保持50% ~ 60%的1-辛烯选择性. Kuhlmann等[67]研究了将环烷基引入PNP配体的N-取代基对Cr-PNP体系催化性能的影响, 引入环庚基的Cr-PNP体系经MMAO-3A活化后, 在60 ℃和4.5 MPa反应条件下催化乙烯四聚生成67.4%的1-辛烯, 反应活性为737 536 g/(gCr·h)(图 16). Blann等[33]的实验表明向苯环中引入两个乙基取代基, 体系的1-己烯和1-辛烯选择性分别为55.6%和34.1%; 而减少一个乙基取代基, 体系的1-辛烯选择性有显著提高(1-己烯: 19.5%, 1-辛烯: 62.1%). 一般来说, 较小的铬中心空间位阻有利于1-辛烯的生成, 而1-己烯形成于更拥挤的铬中心. Overett等[34]研究了苯环取代基对Cr-PNP乙烯四聚催化剂选择性的影响, 向苯环取代基的邻位引入一个甲氧基基团会使体系由四聚催化剂转变为三聚催化剂(1-己烯: 61.7%, 1-辛烯: 16.7%); 然而引入一个相同体积的乙基基团, 体系的1-己烯和1-辛烯的选择性未见明显变化. 显然, Cr系乙烯齐聚催化剂的选择性受到多种因素的共同影响, 某种单一因素并不能用于预测体系的选择性.

|
图 16 改性的Cr-PNP乙烯选择性四聚催化体系 Fig.16 Modified Cr-PNP catalytic system for ethylene tetramerization |
由于MAO的价格较高, 研究人员尝试采用简单易得的助催化剂来取代MAO助催化剂. 早在2007年, McGuinness等[29-30]就使用Cr-PNP体系研究了各种助催化剂的影响, 发现使用氟化硼烷和硼酸盐助催化剂与烷基化试剂结合得到的Cr-PNP催化剂的选择性与使用MAO的Cr-PNP体系相当. 使用[Ph3C][Al(OC(CF3)3)4]和AlEt3作为Cr-PNP体系的助催化剂, 在45 ℃和4.0 MPa反应条件下获得了71.5%的1-辛烯选择性, 催化活性为125 600 g/(gCr·h)[30]. 近年来, Lee等[75-78]使用阴离子[B(C6F5)4]-与阳离子[Cr-PNP]+配合物合成的催化剂经烷基铝助剂活化后可催化乙烯四聚生成1-辛烯, 并且所得1-辛烯的选择性与使用MAO的Cr-PNP催化体系相当. 一般认为MAO最重要的作用是与铬源、配体作用形成乙烯齐聚活性中心. Hirscher等[79]以芳基铬(Ⅲ)配合物为前体, 与PNP配体和HBAr′4反应后用于催化乙烯齐聚. 该体系不使用任何烷基铝助剂, 并且能够达到Cr-PNP/MAO体系的1-己烯/1-辛烯选择性.
2.2 Cr-PCCP体系除了PNP配体外, 萨索尔公司还研究了其他双磷配体. 与PNP配体类似的PCP配体的性能较差[80-81], 而PCCP配体则表现出较好的选择性和活性. 使用PCCP配体合成的Cr-PCCP配合物催化乙烯齐聚可以达到80%的1-己烯和1-辛烯总选择性(1-辛烯选择性约为60%), 活性最高可达7 200 000 g/(gCr·h)[80]. Cr-PCCP/MMAO-3A催化体系, 采用具有刚性芳香基骨架的PCCP配体, 在60 ℃和5.0 MPa反应条件下获得了56.8%的1-辛烯和13.0%的1-己烯, 反应活性为2 240 000 g/(gCr·h)(图 17)[80].

|
图 17 Cr-PCCP乙烯选择性四聚催化体系 Fig.17 Cr-PCCP catalytic system for ethylene tetramerization |
随后, 研究人员还探索了碳桥长度、饱和度、取代基等条件对乙烯齐聚活性和选择性的影响[80, 82-87]. SK能源(SK Energy)的研究人员使用双(二苯基膦基-1, 2-二甲基乙烷)配体与CrCl3(THF)3合成的Cr-PCCP配合物, 经MMAO-12A活化后, 在25 ℃和3.0 MPa的反应条件下获得了77.4%的1-辛烯和9.4%的1-己烯, 反应活性为580 005 g/(gCr·h)(原文为30 158 kg/(molCr·h), 图 18)[82].

|
图 18 Cr-PCCP乙烯选择性四聚催化体系 Fig.18 Cr-PCCP catalytic system for ethylene tetramerization |
Shaikh等[88]使用Ph2PN(R)(CH2)nN(R)PPh2(n = 3, R = Me)二齿配体和CrCl2(THF)2合成的Cr-PNC3NP配合物经DMAO活化后, 在80 ℃和4.0 MPa反应条件下获得了93.1%(原文为91%(摩尔分数))的高1-辛烯选择性, 反应活性为1833 g/(gCr·h)(图 19). 且除1-己烯外该体系不产生聚乙烯等副产物. 对反应条件优化后, 以CrCl3(THF)3为铬源、以DMAO/Et3Al为活化剂, 反应活性提高了近14倍, 1-辛烯选择性略微降低, 但同时也生成了4.1 g聚乙烯. 随后, Gao等[89]使用1, 4-双(二苯基膦)哌嗪配体与CrCl3(THF)3合成的配合物催化乙烯得到分布较宽的α-烯烃.

|
图 19 Cr-PNC3NP乙烯选择性四聚催化体系 Fig.19 Cr-PNC3NP catalytic system for ethylene tetramerization |
Yang等[90]使用混合N, P-配体、Cr(acac)3和MAO原位生成的催化剂进行乙烯低聚实验, 发现使用二苯基磷吡啶配体合成的Cr-PN配合物, 在40 ℃和5.0 MPa反应条件下进行乙烯低聚反应时, 1-辛烯选择性可达73.7%, 反应活性为16 309 g/(gCr·h)(原文为848 g/(mmolCr·h), 图 20). 通过调整反应条件(温度、乙烯压力、催化剂负载量和配体/Cr比), 可以调节1-己烯/1-辛烯的摩尔比在0.3~20之间变化.

|
图 20 Cr-PN乙烯选择性四聚催化体系 Fig.20 Cr-PN catalytic system for ethylene tetramerization |
Zhang等[91]使用Ph2PCH2Si(CH3)2CH2PPh2硅桥连配体与CrCl3(THF)3合成了一种Cr-PCSiCP配合物, 经DMAO/AlEt3活化后, 在0 ℃和1.0 MPa反应条件下获得了86.4%的1-辛烯, 催化活性仅为217 g/(gCr·h)(原文为11 300 g/(molCr·h), 图 21). 而降低铬源用量并在45 ℃和4.0 MPa反应条件下, 1-辛烯选择性降低至78.4%, 催化活性提高至80 975 g/(gCr·h)(原文为4 210 400 g/(molCr·h)).

|
图 21 Cr-PCSiCP乙烯选择性四聚催化体系 Fig.21 Cr-PCSiCP catalytic system for ethylene tetramerization |
通过改变配体的骨架结构对Cr-PCSiCP体系进行调整, 发现使用硅桥连的PCSiNP配体合成的配合物经DMAO/AlEt3活化后, 在15 ℃和4.0 MPa反应条件下获得了83.3%的1-辛烯, 体系的催化活性为43 461 g/(gCr·h)(图 22)[92].

|
图 22 Cr-PCSiNP乙烯选择性四聚催化体系 Fig.22 Cr-PCSiNP catalytic system for ethylene tetramerization |
Alam等[93]使用Si桥连的PPP配体合成的配合物经DMAO/AlEt3活化后, 在45 ℃和4.0 MPa反应条件下获得了71.0%的1-辛烯, 体系的催化活性为3173 g/(gCr·h)(图 23). 而采用减少一个连接Si和P的CH2碳桥的配体也能达到72.0%的1-辛烯选择性.

|
图 23 Cr-(Si)PPP乙烯选择性四聚催化体系 Fig.23 Cr-(Si)PPP catalytic system for ethylene tetramerization |
Radcliffe等[94]使用1-膦基甲亚胺配体(iPr)2P-C(Ph)=N(o-Xyl)与Cr(EH)3合成的一种Cr-PCN配合物经MMAO-3A活化后, 以ZnEt2为助剂, 在60 ℃、4.0 MPa乙烯压力的反应条件下获得了96.7%的总1-己烯和1-辛烯选择性(1-己烯= 35.6%, 1-辛烯= 61.1%), 反应活性为491 000 g/(gCr·h)(图 24).

|
图 24 Cr-PCN乙烯选择性四聚催化体系 Fig.24 Cr-PCN catalytic system for ethylene tetramerization |
表 2中按1-辛烯的选择性从高到低依次列出了典型的高选择性乙烯四聚催化体系. 其中1-辛烯选择性最高可达93.1%, 但该条件下活性偏低, 大多数活性较高的乙烯四聚催化体系的1-辛烯选择性在50%~80%之间. 乙烯四聚催化体系的主要产物还包括1-己烯, 两种产物的总选择性最高可达95%以上. Cr-PNP催化体系也是乙烯四聚催化体系中研究最为广泛的一类乙烯四聚体系, 经过配体的设计可以调控Cr-PNP催化体系的选择性在乙烯三聚和乙烯四聚之间转换.
| 表 2 典型的铬系乙烯四聚催化体系 Table 2 Chromium-based catalysts for ethylene tetramerization. |
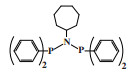
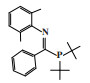
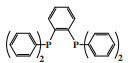
1964年, Cossee和Arlman提出Ziegler-Natta催化剂催化烯烃聚合反应机理, 该机理认为活性中心Ti上存在一个空位, 烯烃分子在空位与Ti中心配位后插入Ti烷基键进行链增长反应, 链终止则通过β-H向Ti中心或者单体转移进行[95-96]. 目前, Cossee-Arlman机理(图 25)也通常被用来解释金属催化剂催化烯烃聚合及齐聚反应, 其主要步骤包括: (1)金属催化剂前体与助催化剂反应生成金属氢或者金属烷基活性中心; (2)一分子乙烯配位到金属活性中心的空位点; (3)乙烯插入金属-氢键或者金属-烷基键实现一个链节的增长, 并形成新的空位点; (4)乙烯配位到金属中心并继续插入金属-碳键进行链增长; (5)通过β-H消除释放相应的α-烯烃, 实现金属活性中心的再生. 遵循Cossee-Arlman机理的催化体系的选择性取决于烯烃单体的插入速率与β-H消除速率的差异, 若插入速率与消除速率相当则体系生成具有统计学分布的α-烯烃.

|
图 25 乙烯聚合、齐聚的Cossee-Arlman反应机理 Fig.25 Proposed Cossee-Arlman mechanism of ethylene polymerization/oligomerization |
1977年, Manyik等[97]首先提出在乙烯三聚成1-己烯的过程中存在铬环戊烷中间体. 后来, Briggs等[98]提出在乙烯三聚过程中, 关键中间体铬环戊烷进一步扩环为铬环庚烷, 整个催化循环包括: (1)两个乙烯分子与铬中心配位; (2)配位乙烯氧化耦合产生铬环戊烷中间体; (3)第三分子乙烯插入铬环戊烷, 扩环形成铬环庚烷; (4)铬环庚烷经历β-H转移释放1-己烯(图 26). 金属环机理比传统的Cossee-Arlman聚合机理更能合理地解释乙烯三聚反应对1-己烯的高选择性. 根据金属环机理, 金属环庚烷中间体的扩环反应和β-H转移反应的竞争决定了体系1-己烯的选择性.

|
图 26 乙烯三聚金属环机理 Fig.26 Proposed metallacycle mechanism of ethylene trimerization |
关键金属环中间体的分解经历一步路径或两步路径(图 27). 一步路径即协同的β-H转移, β-H在铬中心的协同作用下直接转移至α’-C, 然后发生还原消除得到α-烯烃. 两步路径中, β-H先转移至金属中心形成金属烯基氢配合物, 再发生还原消除生成α-烯烃. 由于金属五元环结构中的几何限制, 理论计算中无法定位到金属环戊烷通过协同的β-H转移进行环分解的过渡态, 因此金属五元环中间体遵循两步法氢转移进行开环反应[45, 99-100]. 而金属环庚烷通过一步路径进行环分解的能垒低于两步路径的环分解能垒, 表明1-己烯经协同的β-H转移路径生成[27, 50, 99-103].

|
图 27 以铬环戊烷为例的金属环中间体分解的一步路径和两步路径 Fig.27 The decomposition of the key metallacycle intermediates undergoes a one-step process or follows a two-step pathway |
金属环机理得到了氘同位素标记等实验的支持[104-110]. Emrich等[104]使用(Me2NC2H4C5Me4)Cr(Cl)2与乙烯反应获得铬环戊烷和铬环庚烷, 且铬环庚烷分解释放1-己烯. 在乙烯和过量三甲基膦存在下, 用活性Mg还原CpCr(THF)Cl2获得了单乙烯铬配合物(η2-CH2=CH2)Cr. 最近, Chabbra等[108]通过原位电子顺磁共振光谱与催化剂前体和底物的选择性同位素标记, 在实验中观察到了Cr(Ⅰ)双乙烯物种([Cr-(C2H4)2(CO)2(PNP)]+). 氘同位素标记实验是以等摩尔混合的C2D2和C2H2进行乙烯聚合实验, 若遵循金属环机理则会获得C6D12、C6D8H4、C6D4H8、C6H12; 若遵循Cossee-Arlman机理则会获得H或D数量为奇数的同位素异构体. Agapie等[105]以1∶1混合的C2D2和C2H2为原料, 采用合成的Cr-PNPOMe催化剂进行多次实验, 均获得了比例为1∶3∶3∶1的同位素异构体C6D12、C6D8H4、C6D4H8、C6H12. 以上实验结果均支持乙烯三聚生成1-己烯遵循金属环反应机理.
3.2.2 乙烯四聚金属环机理Overett等[106]使用Cr-PN(iPr)P乙烯四聚催化剂进行氘同位素标记实验, 其结果也支持用金属环机理来解释乙烯四聚反应. 乙烯四聚金属环机理从铬环庚烷中间体开始: (1)第四分子乙烯配位到铬环庚烷中间体的铬中心; (2)乙烯迁移插入铬环庚烷中间体形成铬环壬烷中间体; (3)铬环壬烷中间体通过一步法β-H转移释放1-辛烯(图 28). 根据金属环机理, 铬环庚烷中间体进行环增长与环分解反应之间的竞争决定了体系1-辛烯的选择性.

|
图 28 乙烯四聚金属环机理 Fig.28 Proposed metallacycle mechanism of ethylene tetramerization |
Agapie等[105]针对Cr-PNPOMe催化剂催化乙烯三聚的实验研究发现, 采用NaB[C6H3(CF3)2]4活化后的Cr-PNPOMe催化体系表现出较高的乙烯三聚活性, 这一实验现象表明乙烯三聚催化体系的活性中心为铬阳离子中心. Qi等[100]基于菲利普斯催化体系设计了10种可能的活性中心模型, 使用密度泛函理论研究了菲利普斯催化体系中DMP在乙烯聚合反应中的作用, 发现阳离子模型的计算结果与实验结果相符. 对于金属环机理中铬氧化态的问题, 大多数实验和计算研究都支持乙烯三聚/四聚催化体系的铬中心遵循Cr(Ⅰ)/Cr(Ⅲ)循环[20, 27, 44, 102-103, 111-115]. Yang等[20]研究了菲利普斯体系催化乙烯三聚的金属环机理, 发现Cr(Ⅰ)/Cr(Ⅲ)循环更能合理解释该体系的1-己烯选择性. Rucklidge等[111]使用PNP-型配体制备了Cr(0)和Cr(Ⅰ)羰基配合物进行乙烯齐聚实验, 发现Cr(Ⅰ)配合物/烷基铝可以催化乙烯四聚获得1-辛烯, 而Cr(0)配合物在相同反应条件下无活性. 这一发现支持了Cr(Ⅰ)和Cr(Ⅲ)催化循环. 针对Cr-SNS体系, Yang等[102]研究了金属活性中心的氧化态, 计算表明六重态的双乙烯配位的Cr(Ⅰ)物种进行氧化耦合形成铬环戊烷之前会经过最小能量交叉点(MECP)转变为四重态基态. 双乙烯配位的Cr(Ⅰ)物种从六重态到四重态的自旋交叉降低了形成铬环戊烷的能垒, 表明乙烯三聚反应路径上各中间体和过渡态的铬中心氧化态在Cr(Ⅰ)和Cr(Ⅲ)之间变化. 此外, Monillas等[112]使用合成的Cr(Ⅰ)-N, N配合物在不加助催化剂的条件下催化乙烯三聚生成1-己烯, 并且发现单核Cr(Ⅲ)环戊二烯与乙烯聚合也能生成1-己烯, 这些结果同样支持Cr(Ⅰ)/Cr(Ⅲ)循环.
采用DFT计算方法研究铬中心的氧化态和自旋态, 其计算结果通常取决于计算水平的选择. McGuinness等[44]针对Cr-PNP催化剂开展了基于不同理论方法的基准测试, 发现M06L泛函比较适用于铬系催化乙烯齐聚反应的计算. 作者使用M06L泛函的计算结果表明Cr(Ⅰ)/Cr(Ⅲ)循环比Cr(Ⅱ)/Cr(Ⅳ)循环更可能发生, 并且催化循环中包含从六重态到四重态的自旋交叉反应. 此外, Cr(Ⅱ)/Cr(Ⅳ)循环也得到了一些研究者的支持[41, 116-118]. Rensburg等[117]使用DFT方法研究了菲利普斯催化剂催化乙烯三聚的反应机理, 计算结果表明Cr(Ⅱ)和Cr(Ⅳ)物种的基态是三重态, 并且该催化循环中无自旋交叉发生. Bhaduri等[118]使用DFT方法研究了中性和阳离子物种的催化循环, 发现阳离子[Cr(Ⅱ)Cl2]+模型的Cr(Ⅱ)/Cr(Ⅳ)金属环路径是最有利的路径, 并且所有结构表现为单重态基态. 与Cr(Ⅰ)/Cr(Ⅲ)催化循环相比, Cr(Ⅱ)/Cr(Ⅳ)催化循环并未得到学术界的一致认可.
3.2.4 乙烯三聚反应中的C10副产物Cr-PNPOMe催化剂催化乙烯三聚可以生成高达90%的1-己烯, 该反应还生成10%左右的C10副产物[25]. Do等[119]使用Cr-PNPOMe催化剂进行乙烯和α-烯烃的共齐聚来研究乙烯三聚反应中C10副产物的形成机理. 实验结果表明, Cr-PNPOMe催化乙烯和1-己烯聚合可以生成94%的C10产物. 作者使用GC/MS分析了C10产物并提出C10产物的形成路径(图 29), 主要步骤包括: (1)乙烯配位并插入铬环戊烷生成铬环庚烷, 随后释放1-己烯; (2)1-己烯配位并以2, 1-插入铬环戊烷或者乙烯从低位阻Cr-C键一侧进行配位并插入2-丁基铬环戊烷后, 经β-H消除生成癸烯; (3)1-己烯配位并以1, 2-插入铬环戊烷, 或者乙烯配位并插入2-丁基铬环戊烷或者3-丁基铬环戊烷后, 经β-H消除生成含有支链的C10烯烃. 作者认为乙烯三聚生成1-己烯与乙烯/1-己烯共聚相互竞争. Zilbershtein等[120]使用菲利普斯催化体系研究了乙烯三聚反应中副产物癸烯的形成, 作者通过NMR和GC/MS确定反应产物中存在图 29中所预期的乙烯与1-己烯共三聚生成的癸烯和支链烯烃, 该研究结果支持上述C10副产物的形成路径. 此外, Gong等[48]通过DFT方法研究了Cr-PNPOMe体系的乙烯与1-己烯共三聚机理, 计算得到的C10产物分布与实验结果一致.

|
图 29 乙烯三聚反应的C10副产物的生成路径 Fig.29 C10 Alkenes formed via co-trimerization of ethylene and 1-hexene |
Overett等[106]使用Cr-PNP/MMAO体系催化乙烯四聚除获得了70%左右的1-辛烯外, 还发现以1∶1的比例形成的甲基环戊烷和亚甲基环戊烷两种副产物. 作者提出铬环庚烷重排为铬甲基环戊基氢配合物主要有协同反应和分步反应(包括β-H转移和插入成环)两种途径. 铬甲基环戊基氢配合物生成甲基环戊烷和亚甲基环戊烷也可能存在两种方式: (1)还原消除产生甲基环戊烷或β-H转移生成亚甲基环戊烷; (2)通过双金属歧化机理生成两种环状副产物(图 30). 作者认为双核机理可以解释实验中观察到的1∶1比例的甲基环戊烷和亚甲基环戊烷. Britovsek等[45]使用DFT方法研究了Cr-PNP体系, 计算发现乙基己烯基铬中间体通过己烯基尾部插入形成乙基甲基环戊基铬中间体的能垒低于通过C-C还原消除形成1-辛烯和β-H反向转移形成1-己烯的能垒, 且乙基甲基环戊基铬中间体的β-H转移生成甲基环戊烷和亚甲基环戊烷的能垒仅差5.9 kJ/mol, 符合实验观察到的1∶1的产率. Zhang等[91]在Cr/硅桥连的双磷配体体系催化的乙烯聚合反应的产物中观察到了非1∶1比例的甲基环戊烷和亚甲基环戊烷, 作者认为双金属歧化机理不能解释此现象, 且形成甲基环戊烷和亚甲基环戊烷的活性中心可能与乙烯齐聚活性中心不同. Boelter等[86]采用多种双磷配体进行乙烯齐聚的实验结果表明, 甲基环戊烷与亚甲基环戊烷的比率随配体的改变在1.25~5.00之间变动, 因此作者支持通过还原消除产生甲基环戊烷或β-H转移生成亚甲基环戊烷.

|
图 30 副产物甲基环戊烷和亚甲基环戊烷的可能路径 Fig.30 Proposed mechanisms for the formation of methylcyclopentane and methylenecyclopentane |
Wilke等[121]提出炔烃选择性四聚生成环辛四烯路径中存在双金属环戊二烯中间体. 在此启发下, Peitz等[122]提出了乙烯四聚双金属机理. 乙烯四聚双金属机理的关键中间体是双金属铬环戊烷, 其反应步骤包括: (1)双金属配合物吸附4分子乙烯配位到两个金属中心; (2)随后两个金属中心上的两分子乙烯氧化耦合形成两个金属环戊烷; (3)两个五元环单元C-C偶联形成更大的双金属十元环; (4)最后双金属十元环通过β-H转移释放1-辛烯(图 31). 双金属机理避开了1-己烯的形成, 而直接经由双金属十元环中间体释放1-辛烯. 乙烯四聚双金属机理将乙烯三聚和四聚催化体系之间的选择性差异归因于铬源与配体之间形成双金属配合物的能力.

|
图 31 乙烯四聚双金属机理 Fig.31 Proposed binuclear mechanism for ethylene tetramerization |
Hirscher等[110]通过同位素标记实验对乙烯四聚双金属机理进行了评估. 作者认为在乙烯双金属机理中, 选择性由金属环戊烷的C-C偶联反应与乙烯插入的相对速率决定. 两者都不涉及C-H或者Cr-H键的形成或裂解, 因此不存在一级同位素效应. 然而实验获得的KIE=3.0 ± 0.3(同位素效应)表明该反应是一级同位素效应控制的反应. 该实验结果并不支持乙烯四聚双金属机理.
Britovsek等[45]提出双配位机理来解释Cr-PNP催化体系催化乙烯齐聚中1-辛烯的形成(图 32). 双配位机理反应路径包括: (1)一分子乙烯在铬环戊烷上进行配位(单配位)后形成单乙烯铬环戊烷中间体, 随后经过迁移插入形成铬环庚烷, 再通过β-H还原消除释放1-己烯; (2)两分子乙烯在铬环戊烷中间体上进行配位(双配位)形成双乙烯铬环戊烷中间体, 随后经过两分子配位乙烯的连续迁移插入形成铬环壬烷, 再经过还原消除生成1-辛烯. 双配位机理中反应经双乙烯铬环戊烷中间体形成1-辛烯, 而1-己烯主要经单乙烯铬环戊烷中间体形成. 催化反应的选择性取决于单配位路径和双配位路径之间的竞争.

|
图 32 乙烯四聚双配位机理 Fig.32 Proposed double-coordination mechanism for ethylene tetramerization |
自2002年以来, 工业界和学术界对铬系乙烯选择性三聚、四聚催化体系的研发热度持续增加. 乙烯选择性三聚催化剂, 如菲利普斯铬系催化体系、Cr-PNP体系、Cr-SNS体系等, 对1-己烯的选择性可达99.9%. 乙烯四聚催化剂, 如Cr-PNP体系、Cr-PCCP体系等, 对1-辛烯选择性普遍在50% ~ 80%之间. 调控体系的1-己烯、1-辛烯选择性也是设计开发高效乙烯三聚/四聚反应催化剂的一大难点, 尤其是提高1-辛烯的选择性. Cr(EH)3/DMP/二乙基氯化铝/三乙基铝乙烯三聚催化体系早在2003年就由雪佛龙菲利普斯化工公司成功实现工业化生产. 2014年, 萨索尔公司采用Cr-PNP催化体系进行乙烯四聚的工业化试验. 国内大庆石化公司日前已完成5000 t/a 1-己烯的工业化试验, 其乙烯四聚工业试验项目也在建设中. 乙烯三聚/四聚工业化面临的另一个难题是减少聚乙烯的形成, 但目前对乙烯三聚/四聚反应中聚乙烯的形成机理尚未有定论[123-124].为了加快乙烯三聚/四聚催化剂的工业化应用, 我们仍需进一步对乙烯齐聚的反应机理、活性中心、金属氧化态以及助催化剂等方面进行更为深入的研究.
乙烯选择性三聚/四聚领域仍存在诸多挑战与机遇. 一方面, 随着现代实验表征技术的发展, 科研工作者可以原位追踪催化活性中心上发生的反应动态; 另一方面, 高性能计算机和算法的发展也促进了以机器学习和密度泛函理论为代表的数学统计和分子模拟方法在新型催化剂筛选和设计领域的应用. 通过结合实验表征和理论计算对催化反应机理进行精准解析, 必将加速新型催化剂的开发进程, 最终实现乙烯高选择性三聚、四聚的工业规模生产.
| [1] |
Guo Feng(郭峰), Li Chuan-feng(李传峰), Liu Jing-wei(刘经伟), et al. Processing technology and technique progress in linear α-olefins(线性α-烯烃的生产工艺和技术进展)[J]. Mod Chem Ind(现代化工), 2010, 30(S2): 92-96.
|
| [2] |
a. Alferov K A, Babenko I A, Belov G P. New catalytic systemson the basis of chromium compounds for selective synthe-sis of 1-hexene and 1-octene[J]. Petro Chem, 2017, 57(1): 1-30. b. Wang Jing(王静), Shi Yong-sen(史永森), Li Ya-ling(李亚玲), et al. The development of catalysts for polyolefin-based elastomer(聚烯烃弹性体催化剂研究进展) [J]. J Mol Catal(China)(分子催化), 2020, 34(6): 579-591. |
| [3] |
Alferov K A, Belov G P, Meng Y. Chromium catalysts forselective ethylene oligomerization to 1-hexene and 1-octene: Recent results[J]. Appl Catal, A, 2017, 542: 71-124.
|
| [4] |
Feng Zhi-chao(封智超), Mao Guo-liang(毛国梁), Wu Wei(吴韦), et al. Research progress of chromium-basedcatalysts for selective polymerization of ethylene(铬系乙烯选择性齐聚催化剂的研究进展)[J]. Acta Petro Sin(Petro Process Sect)(石油学报(石油加工)), 2018, 34(1): 190-202.
|
| [5] |
Wei Xue-ying(魏雪莹), Wu Wei(吴玮), Nai Yong-ning(乃永宁), et al. Research progress on the selective oligom-erization of ethylene catalyzed by phosphoramine chromiumand diphosphinoamine chromium(膦胺铬和双膦铬催化乙烯选择性齐聚研究进展)[J]. Chin J Appl Chem(应用化学), 2021, 38(2): 136-156.
|
| [6] |
Jie Su-yun(介素云), Zhang Shu(张树), Sun wen-hua(孙文华). Progress on research of catalysts for ethylene oligom-erization(乙烯齐聚催化剂的研究进展)[J]. PetrochemTechnol(石油化工), 2006, 35(3): 295-300.
|
| [7] |
Cao Jun(曹军), Zhang Bao-jun(张宝军), Li Ming-yuan(李明远), et al. Progress in research of ethylene oligomeriz-ation by chromium-based homogeneous catalyst(乙烯齐聚均相铬系催化剂的研究进展)[J]. Petrochem Technol(石油化工), 2007, 36(3): 304-309.
|
| [8] |
Liu Z, He X, Cheng R, et al. Chromium catalysts for ethy-lene polymerization and oligomerization[J]. Adv Chem Eng, 2014, 44: 127–191.
|
| [9] |
Liu Rui(刘睿), Xiao Shu-meng(肖树萌), Zhong Xiang-hong(钟向宏), et al. Advances in selective ethylene oligomeriz-ation based on [PNP]-ligand chromium catalysts(基于[PNP]配体的铬催化剂体系选择性催化乙烯齐聚的研究进展)[J]. Chin J Org Chem(有机化学), 2015, 35(9): 1861-1888.
|
| [10] |
Petit J, Magna L, Mezailles N. Alkene oligomerization via metallacycles: Recent advances and mechanistic insights[J]. Coord Chem Rev, 2022, 450: 1–16.
|
| [11] |
Tembe G. Catalytic tri- and tetramerization of ethylene: A mechanistic overview[J]. Catal Rev, 2022, 2022: 1–56.
|
| [12] |
Reagen W K. Process for olefin polymerization[P]. EP: 0417477, 1991.
|
| [13] |
Freeman J W, Buster J L, Knudsen R D. Preparation of homogeneous catalysts for olefin oligomerization[P]. US: 5856257, 1999.
|
| [14] |
Araki Y, Nakamura H, Nanba Y, et al. Process for produ-cing α-olefin oligomer[P]. US: 5856612, 1999.
|
| [15] |
Yang Y, Kim H, Lee J, et al. Roles of chloro compo-und in homogeneous [Cr(2-ethylhexanoate)3/2, 5-dim-ethylpyrrole/triethylaluminum/chloro compound] catalyst system for ethylene trimerization[J]. Appl Catal, A: Gen, 2000, 193(1/2): 29-38.
|
| [16] |
Tang S, Liu Z, Yan X, et al. Kinetic studies on the pyrrole-Cr-based Chevron-Phillips ethylene trimerization catalyst system[J]. Appl Catal, A: Gen, 2014, 481: 39-48.
|
| [17] |
Luo H K, Li D G, Li S. The effect of halide and the coor-dination geometry of chromium center in homogeneous catalyst system for ethylene trimerization[J]. J Mol Catal A: Chem, 2004, 221(1/2): 9–17.
|
| [18] |
Jiang T, Ji R, Chen H, et al. Effect of alkylaluminum activators on ethylene trimerization based on 2, 5-DMP/Cr(Ⅲ)/TCE catalyst system[J]. Chin J Chem, 2011, 29(6): 1149–1153.
DOI:10.1002/cjoc.201190215 |
| [19] |
Bahri-Laleh N, Karimi M, Kalantari Z, et al. H2 effect in Chevron-Phillips ethylene trimerization catalytic system: An experimental and theoretical investigation[J]. Polym Bull, 2018, 75(8): 3555–3565.
DOI:10.1007/s00289-017-2228-3 |
| [20] |
Yang Y, Liu Z, Cheng R, et al. Mechanistic DFT study on ethylene trimerization of chromium catalysts suppo-rted by a versatile pyrrole ligand system[J]. Organome-tallics, 2014, 33(10): 2599–2607.
DOI:10.1021/om500306a |
| [21] |
Naji-Rad E, Gimferrer M, Bahri-Laleh N, et al. Explor-ing basic components effect on the catalytic efficiency of Chevron-Phillips catalyst in ethylene trimerization[J]. Catalysts, 2018, 8(6): 1–11.
|
| [22] |
Yoshida T, Yamamoto T, Okada H, et al. Catalyst for trimerization of ethylene and process for trimerizing ethylene using the catalyst[P]. US: 0035029, 2002.
|
| [23] |
Li Ping(李平), Mi Pu-ke(米普科), Gu Jin-jing(顾瑾璟), et al. Synthesis of novel homogeneous chromium-based catalysts and catalyze ethylene oligomerization(新型均相铬系催化剂的制备与催化乙烯齐聚)[J]. Chem J Chin Univer(高等学校化学学报), 2017, 38(11): 2111-2117.
|
| [24] |
Wu F J, La B R. Ethylene trimerization[P]. US: 5811618, 1998.
|
| [25] |
Carter A, Cohen S A, Cooley N A, et al. High activity ethylene trimerization catalysts based on diphosphine ligands[J]. Cheml Commun, 2002, 2002(8): 858–859.
|
| [26] |
McGuinness D S, Wasserscheid P, Keim W, et al. Novel Cr-PNP complexes as catalysts for the trimerization of ethylene[J]. Cheml Commun, 2003, 3(3): 334–335.
|
| [27] |
Klemps C, Payet E, Magna L, et al. PCNCP ligands in the chromium-catalyzed oligomerization of ethylene: Tri- versus tetramerization[J]. Chem-Eur J, 2009, 15(33): 8259–8268.
DOI:10.1002/chem.200900986 |
| [28] |
Agapie T, Day M W, Henling L M, et al. A chromium-diphosphine system for catalytic ethylene trimerization: Synthetic and structural studies of chromium complexes with a nitrogen-bridged diphosphine ligand with ortho-methoxyaryl substituents[J]. Organometallics, 2006, 25(11): 2733–2742.
DOI:10.1021/om050605+ |
| [29] |
McGuinness D S, Overett M, Tooze R P, et al. Ethylene tri- and tetramerization with borate cocatalysts: Effects on activity, selectivity and catalyst degradation pathways[J]. Organometallics, 2007, 26(4): 1108–1111.
DOI:10.1021/om060906z |
| [30] |
McGuinness D S, Rucklidge A J, Tooze R P, et al. Cocatalyst influence in selective oligomerization: Effecton activity, catalyst stability and 1-hexene/1-octeneselectivity in the ethylene trimerization and tetramer-ization reaction[J]. Organometallics, 2007, 26(10): 2561–2569.
DOI:10.1021/om070029c |
| [31] |
Lifschitz A M, Hirscher N A, Lee H B, et al. Ethylene tetramerization catalysis: Effects of aluminum-indu-ced isomerization of PNP to PPN ligands[J]. Organom-etallics, 2017, 36(8): 1640–1648.
DOI:10.1021/acs.organomet.7b00189 |
| [32] |
Grauke R, Schepper R, Rabeah J, et al. Impact of Al activators on structure and catalytic performance of Cr catalysts in homogeneous ethylene oligomerization-A multitechnique in situ/operando study[J]. ChemCat-Chem, 2019, 12(4): 1025–1035.
|
| [33] |
Blann K, Bollmann A, Dixon J T, et al. Highly selective chromium-based ethylene trimerisation catalysts with bulky diphosphinoamine ligands[J]. Cheml Commun, 2005, 2005(5): 620–621.
|
| [34] |
Overett M J, Blann K, Bollmann A, et al. Ethylene trimerisation and tetramerisation catalysts with polar-substituted diphosphinoamine ligands[J]. Cheml Commun, 2005, 2005(5): 622–624.
|
| [35] |
Li Bo-min(李博民), Liu Bo-ping(刘柏平), Liu Zhen(刘振). Ethylene selective tri-/tetramerisation with PNP-Cr mixed ligands(PNP-Cr复合配体用于乙烯选择性三聚/四聚的研究)[J]. J East China Univ Sci Technol(Nat Sci Ed)(华东理工大学学报(自然科学版)), 2018, 44(3): 303-307.
|
| [36] |
Jiang Meng-yuan(姜梦圆), Mao Guo-liang(毛国梁), TianShi-wei(田诗伟), et al. Advances in multi-site PNP ligandsfor selective oligomerization of ethylene(多位点PNP配体在铬催化乙烯选择性齐聚中的应用进展)[J]. Polym Mater Sci Eng(高分子材料科学与工程), 2020, 36(6): 162-176.
|
| [37] |
Alam F, Fan H, Dong C, et al. Chromium catalysts stabilized by alkylphosphanyl PNP ligands for selective ethylene tri-/tetramerization[J]. J Catal, 2021, 404: 163–173.
DOI:10.1016/j.jcat.2021.09.025 |
| [38] |
Zhang J, Alam F, Fan H, et al. Chromium catalysts based on PNP(NR2)2 ligands for selective ethylene oligomerization[J]. Appl Org Chem, 2021, 36(1): 1–10.
|
| [39] |
Zhang Na(张娜), Li Yu-ying(李昱颖), Tang Jing(唐静), et al. Synthesis and catalytic performance in ethylene oligomerization of mixture metal organic frameworks graft PNP chromium catalyst(MixMOFs接枝PNP铬系催化剂的合成及催化乙烯齐聚性能)[J]. Chemistry(化学通报), 2021, 84(5): 474-479.
|
| [40] |
Tang S, Liu Z, Zhan X, et al. 2D-QSPR/DFT studies of aryl-substituted PNP-Cr-based catalyst systems for highly selective ethylene oligomerization[J]. J Mol Model, 2014, 20(3): 1–13.
|
| [41] |
McGuinness D S, Brown D B, Tooze R P, et al. Ethylene trimerization with Cr-PNP and Cr-SNS complexes: Effect of ligand structure, metal oxidation state and role of activator on catalysis[J]. Organometallics, 2006, 25(15): 3605–3610.
DOI:10.1021/om0601091 |
| [42] |
Schofer S J, Day M W, Henling L M, et al. Ethylene trimerization catalysts based on chromium complexes with a nitrogen-bridged diphosphine ligand having ortho-methoxyaryl or ortho-thiomethoxy substituents: Well-defined catalyst precursors and investigations of the mechanism[J]. Organometallics, 2006, 25(11): 2743–2749.
DOI:10.1021/om0506062 |
| [43] |
Do L H, Labinger J A, Bercaw J E. Spectral studies of a Cr(PNP)-MAO system for selective ethylene trimerization catalysis: Searching for the active species[J]. ACS Catal, 2013, 3(11): 2582–2585.
DOI:10.1021/cs400778a |
| [44] |
McGuinness D S, Chan B, Britovsek G J P, et al. Ethyl-ene trimerisation with Cr-PNP catalysts: A theoretical bench-marking study and assessment of catalyst oxidation state[J]. Aust J Chem, 2014, 67(10): 1481–1490.
DOI:10.1071/CH14436 |
| [45] |
Britovsek G J P, McGuinness D S, Wierenga T S, et al. Single- and double-coordination mechanism in ethylene tri- and tetramerization with Cr/PNP catalysts[J]. ACS Catal, 2015, 5(7): 4152–4166.
DOI:10.1021/acscatal.5b00989 |
| [46] |
Britovsek G J P, McGuinness D S. A DFT mechanistic study on ethylene tri- and tetramerization with Cr/PNP catalysts: Single versus double insertion pathways[J]. Chem-Eur J, 2016, 22(47): 1–7.
|
| [47] |
Britovsek G J P, McGuinness D S, Tomov A K. Mecha-nistic study of ethylene tri- and tetramerization with Cr/PNP catalysts: Effects of additional donors[J]. Catal Sci Technol, 2016, 6(23): 8234–8241.
DOI:10.1039/C6CY02112C |
| [48] |
Gong M, Liu Z, Li Y, et al. Selective co-oligomerization of ethylene and 1-hexene by chromium-PNP catalysts: A DFT study[J]. Organometallics, 2016, 35(7): 972–981.
DOI:10.1021/acs.organomet.5b01029 |
| [49] |
Sun Ren(孙任), Liu Lin(刘霖), Cheng Rui-hua(程瑞华), et al. Effects of ether electron donors on Cr/PNP-catalyzed ethylene selective oligomerization(醚类给电子体对Cr/PNP乙烯选择性齐聚体系催化反应性能的影响)[J]. J Mol Catal(分子催化), 2019, 33(2): 103-112.
|
| [50] |
Liu L, Liu Z, Cheng R, et al. Unraveling the effects of H2, N substituents and secondary ligands on Cr/PNP-catalyzed ethylene selective oligomerization[J]. Organom-etallics, 2018, 37(21): 1–8.
|
| [51] |
Zhang Na(张娜), Shang Yi-teng(商益腾), Li Cui-qin(李翠勤), et al. Synthesis and catalytic performance in ethy-lene oligomerization of hyperbranched PNP chromium catalyst(超支化PNP铬系催化剂的合成及对乙烯齐聚的催化性能)[J]. Polym Mater Sci Eng(高分子材料科学与工程), 2018, 34(8): 20-30.
|
| [52] |
Wang Jun(王俊), Liu Jin-yi(刘锦义), Chen Li-duo(陈丽铎), et al. Synthesis and ethylene oligomerization beha-vior of hyperbranched bispyridineimine chromium catalyst(超支化双吡啶亚胺铬催化剂的合成及催化乙烯齐聚性能)[J]. Chin J Appl Chem(应用化学), 2019, 36(7): 773-781.
|
| [53] |
Wang Jun(王俊), Li Yu-ying(李昱颖), Zhang Na(张娜), et al. Synthesis of hyperbranched chromium catalysts and study on ethylene oligomerization properties(超支化铬系催化剂的合成及乙烯齐聚性能研究)[J]. J Mol Catal(分子催化), 2019, 33(5): 429-437.
|
| [54] |
McGuinness D S, Wasserscheid P, Keim W, et al. First Cr(Ⅲ)-SNS complexes and their use as highly efficient catalysts for the trimerization of ethylene to 1-hexene[J]. J Am Chem Soc, 2003, 125(18): 5272–5273.
DOI:10.1021/ja034752f |
| [55] |
Temple C N, Gambarotta S, Korobkov I, et al. New insightinto the role of the metal oxidation state in controlling the selectivity of the Cr-(SNS) ethylene trimerization catalyst[J]. Organometallics, 2007, 26(18): 4598–4603.
DOI:10.1021/om700452u |
| [56] |
Zhang J, Braunstein P, Hor T S A. Highly selective chrom-ium(Ⅲ) ethylene trimerization catalysts with [NON] and [NSN] heteroscorpionate ligands[[J]. Organometallics, 2008, 27(17): 4277–4279.
DOI:10.1021/om8005239 |
| [57] |
Zhang J, Li A, Hor T S A. Ligand effect on ethylene trimerisation with [NNN]-heteroscorpionate pyrazolyl Cr(Ⅲ) catalysts[J]. Dalton Trans, 2009, 2009(42): 9327–9333.
|
| [58] |
Albahily K, Koc E, Al-Baldawi D, et al. Chromium catal-ysts supported by a nonspectator NPN ligand: Isola-tion of single-component chromium polymerization cataly-sts[J]. Angew Chem Int Edit, 2008, 47(31): 5900–5903.
|
| [59] |
Sydora O L, Jones T C, Small B L, et al. Selective ethylene tri-/tetramerization catalysts[J]. ACS Catal, 2012, 2(12): 2452–2455.
DOI:10.1021/cs300488t |
| [60] |
Kwon D H, Maley S M, Stanley J C, et al. Why less coordination provides higher reactivity chromium phosphi-noamidine ethylene trimerization catalysts[J]. ACS Catal, 2020, 10(17): 9674–9683.
DOI:10.1021/acscatal.0c02595 |
| [61] |
Ogawa T, Lindeperg F, Stradiotto M, et al. Chromium N-phosphinoamidine ethylene tri-/tetramerization catalysts: Designing a step change in 1-octene selectivity[J]. J Catal, 2021, 394: 444–450.
DOI:10.1016/j.jcat.2020.10.021 |
| [62] |
Morgan N, Maley S M, Kwon D H, et al. Computational assessment and understanding of C6 product selectivity for chromium phosphinoamidine catalyzed ethylene trimerization[J]. J Org Chem, 2022, 961: 1–7.
|
| [63] |
Peitz S, Peulecke N, Aluri B R, et al. A selective chrom-ium catalyst system for the trimerization of ethene and its coordination chemistry[J]. Eur J Inorg Chem, 2010, 2010(8): 1167–1171.
DOI:10.1002/ejic.201000044 |
| [64] |
Bollmann A, Blann K, Dixon J T, et al. Ethylene tetrame-rization: A new route to produce 1-octene in exceptionally high selectivities[J]. J Am Chem Soc, 2004, 126(45): 14712–14713.
DOI:10.1021/ja045602n |
| [65] |
Blann K, Bollmann A, De Bod H, et al. Ethylene tetramer-ization: Subtle effects exhibited by N-substituted diphosphinoamine ligands[J]. J Catal, 2007, 249(2): 244–249.
DOI:10.1016/j.jcat.2007.04.009 |
| [66] |
Killian E, Blann K, Bollmann A, et al. The use of bis(diphenylphosphino)amines with N-aryl functionalities in selective ethylene tri- and tetramerisation[J]. J Mol Catal A: Chem, 2007, 270(1/2): 214–218.
|
| [67] |
Kuhlmann S, Blann K, Bollmann A, et al. N-substituted diphosphinoamines: Toward rational ligand design for the efficient tetramerization of ethylene[J]. J Catal, 2007, 245(2): 279–284.
DOI:10.1016/j.jcat.2006.10.020 |
| [68] |
Jiang T, Zhang S, Jiang X, et al. The effect of N-aryl bisphosphineamine ligands on the selective ethylene tetramerization[J]. J Mol Catal A: Chem, 2008, 279(1): 90–93.
DOI:10.1016/j.molcata.2007.10.009 |
| [69] |
Cloete N, Visser H G, Engelbrecht I, et al. Ethylene tri- and tetramerization: A steric parameter selectivity switch from X-ray crystallography and computational analysis[J]. Inorg Chem, 2013, 52(5): 2268–2270.
DOI:10.1021/ic302578a |
| [70] |
Jiang T, Ning Y, Zhang B, et al. Preparation of 1-octene by the selective tetramerization of ethylene[J]. J Mol Catal A: Chem, 2006, 259(1/2): 161–165.
|
| [71] |
Chen H, Liu X, Hu W, et al. Effects of halide in homog-eneous Cr(Ⅲ)/PNP/MAO catalytic systems for ethylene tetramerization toward 1-octene[J]. J Mol Catal A: Chem, 2007, 270(1/2): 273–277.
|
| [72] |
Jiang T, Liu X, Ning Y, et al. Performance of various alumi-noxane activators in ethylene tetramerization based on PNP/Cr(Ⅲ) catalyst system[J]. Catal Commun, 2007, 8(7): 1145–1148.
DOI:10.1016/j.catcom.2006.10.032 |
| [73] |
Wang T, Gao X, Shi P, et al. Mixed aluminoxanes: Effic-ient cocatalysts for bisphosphineamine/Cr(Ⅲ) catalyzed ethylene tetramerization toward 1-octene[J]. Appl Petrochem Res, 2015, 5(2): 143–149.
DOI:10.1007/s13203-015-0103-4 |
| [74] |
Makume B F, Holzapfel C W, Maumela M C, et al. Ethyl-ene tetramerisation: A structure-selectivity correlation[J]. ChemPlusChem, 2020, 85(10): 1–11.
|
| [75] |
Kim Eun H, Lee Hyun M, Lee Bun Y, et al. Methylalu-minoxane-free chromium catalytic system for ethylene tetramerization[J]. ACS omega, 2017, 2(3): 765–773.
DOI:10.1021/acsomega.6b00506 |
| [76] |
Kim T H, Lee H M, Park H S, et al. MAO-free and extre-mely active catalytic system for ethylene tetramerization[J]. Appl Org Chem, 2019, 33(4): 1–13.
|
| [77] |
Park H S, Kim T H, Baek J W, et al. Extremely active ethylene tetramerization catalyst avoiding the use of methylalu-minoxane: [iPrN{P(C6H4-p-SiR3)2}2CrCl2]+ [B(C6F5)4][J]. ChemCatChem, 2019, 11(17): 4351–4359.
DOI:10.1002/cctc.201900898 |
| [78] |
Lee J H, Baek J W, Lee D G, et al. Preparation of extre-mely active ethylene tetramerization catalyst [iPrN(PAr2)2-CrCl2]+[B(C6F5)4]- (Ar = -C6H4-p-SiR3)[J]. Catalysts, 2021, 11(9): 1–15.
|
| [79] |
Hirscher N A, Perez S D, Agapie T. Robust chromium prec-ursors for catalysis: Isolation and structure of a single-component ethylene tetramerization precatalyst[J]. J Am Chem Soc, 2019, 141(14): 6022–6029.
DOI:10.1021/jacs.9b01387 |
| [80] |
Overett M J, Blann K, Bollmann A, et al. Carbon-bridged diphosphine ligands for chromium-catalyzed ethylene tetramerization and trimerization reactions[J]. J Mol Catal A: Chem, 2008, 283(1/2): 114–119.
|
| [81] |
Dulai A, de Bod H, Hanton M J, et al. C-substituted bis(diphenylphosphino)methane-type ligands for chromium-catalyzed selective ethylene oligomerization reactions[J]. Organometallics, 2009, 28(15): 4613–4616.
DOI:10.1021/om900285e |
| [82] |
Kim S K, Kim T J, Chung J H, et al. Bimetallic ethylene tetramerization catalysts derived from chiral DPPDME ligands: Syntheses, structural characterizations and catalytic performance of [(DPPDME)CrCl3]2 (DPPDME=S, S- and R, R-chiraphos and meso-achiraphos)[J]. Organometallics, 2010, 29(22): 5805–5811.
DOI:10.1021/om100400b |
| [83] |
Zhang J, Wang X, Zhang X, et al. Switchable ethylene tri-/tetramerization with high activity: Subtle effect presented by backbone-substituent of carbon-bridged diphosphine ligands[J]. ACS Catal, 2013, 3(10): 2311–2317.
DOI:10.1021/cs400651h |
| [84] |
Zheng Ming-fang(郑明芳), Wu Hong-fei(吴红飞), Ma Xu-feng(马旭峰), et al. Ethylene oligomerization cataly-zed by binuclear Cr catalyst based on a bridged phosphine ligand(含桥联多膦配体的双核Cr(Ⅲ)乙烯齐聚催化剂)[J]. Petrochem Technol(石油化工), 2018, 47(9): 924-928.
|
| [85] |
Lee H, Joe Y, Park H. Chromium catalysts for ethylene trimerization/tetramerization functionalized with ortho-fluorinated arylphosphine ligand[J]. Catal Commun, 2019, 121: 15–18.
DOI:10.1016/j.catcom.2018.12.010 |
| [86] |
Boelter S D, Davies D R, Margl P, et al. Phospholane-basedligands for chromium-catalyzed ethylene tri- and tetramer-ization[J]. Organometallics, 2020, 39(7): 976–987.
DOI:10.1021/acs.organomet.9b00722 |
| [87] |
Boelter S D, Davies D R, Milbrandt K A, et al. Evaluation of bis(phosphine) ligands for ethylene oligomerization: Discovery of alkyl phosphines as effective ligands for ethylene tri- and tetramerization[J]. Organo Metallics, 2020, 39(7): 967–975.
DOI:10.1021/acs.organomet.9b00721 |
| [88] |
Shaikh Y, Albahily K, Sutcliffe M, et al. A highly selectiveethylene tetramerization catalyst[J]. Angew Chem Int Edit, 2012, 51(6): 1366–1369.
DOI:10.1002/anie.201106517 |
| [89] |
Gao Xiang-lu(高祥录), Sun Yue-ming(孙月明), Pei Hai-xiang(裴海香), et al. Ethylene oligomerization catalyzed bynovel Cr(Ⅲ) complex/methylaluminoxane(新型Cr(Ⅲ)络合物/甲基铝氧烷催化乙烯齐聚)[J]. Petrochem Technol(石油化工), 2015, 44(2): 181-185.
|
| [90] |
Yang Y, Liu Z, Liu B, et al. Selective ethylene tri-/tetrame-rization by in situ-formed chromium catalysts stabilized by N, P-based ancillary ligand systems[J]. ACS Catal, 2013, 3(10): 2353–2361.
DOI:10.1021/cs4004968 |
| [91] |
Zhang L, Meng X, Chen Y, et al. Chromium-based ethylene tetramerization catalysts supported by silicon-bridged diphosphine ligands: Further combination of high activity and selectivity[J]. ChemCatChem, 2017, 9(1): 76–79.
DOI:10.1002/cctc.201600941 |
| [92] |
Alam F, Zhang L, Wei W, et al. Catalytic systems based on chromium(Ⅲ) silylated-diphosphinoamines for selective ethylene tri-/tetramerization[J]. ACS Catal, 2018, 8(11): 10836–10845.
DOI:10.1021/acscatal.8b02698 |
| [93] |
Alam F, Wang J, Dong C, et al. Chromium(Ⅲ) cataly-sts based on tridentate silicon-bridged tris(diphenyl-phosphine) ligands for selective ethylene tri-/tetrameriz-ation[J]. J Catal, 2020, 392: 278–286.
DOI:10.1016/j.jcat.2020.10.008 |
| [94] |
Radcliffe J E, Batsanov A S, Smith D M, et al. Phosph-anyl methanimine (PCN) ligands for the selective trimeriz-ation/tetramerization of ethylene with chromium[J]. ACS Catal, 2015, 5(12): 7095–7098.
DOI:10.1021/acscatal.5b02106 |
| [95] |
Arlman E J, Cossee P. Ziegler-Natta catalysis Ⅲ. Stereospecific polymerization of propene with the catalyst system TiCl3/AlEt3[J]. J Catal, 1964, 3(1): 99–104.
DOI:10.1016/0021-9517(64)90097-1 |
| [96] |
Cossee P. Ziegler-Natta catalysis I. Mechanism of polymer-ization of α-olefins with Ziegler-Natta catalysts[J]. J Catal, 1964, 3(1): 80–88.
DOI:10.1016/0021-9517(64)90095-8 |
| [97] |
Manyik R M, Walker W E, Wilson T P. A soluble chrom-ium-based catalyst for ethylene trimerization and polymeri-zation[J]. J Catal, 1977, 47(2): 197–209.
DOI:10.1016/0021-9517(77)90167-1 |
| [98] |
Briggs J R. The selective trimerization of ethylene to hex-1-ene[J]. J Chem Soc Chem Commun, 1989, 1989(11): 674–675.
|
| [99] |
Yu Z X, Houk K N. Why trimerization? Computational elucidation of the origin of selective trimerization of ethene catalyzed by [TaCl3(CH3)2] and an agostic-assisted hydride transfer mechanism[J]. Angew Chem Int Edit, 2003, 115(7): 832–835.
DOI:10.1002/ange.200390184 |
| [100] |
Qi Y, Dong Q, Zhong L, et al. Role of 1, 2-dimeth-oxyethane in the transformation from ethylene polymeri-zation to trimerization using chromium tris(2-ethylhex-anoate)-based catalyst system: A DFT study[J]. Organometallics, 2010, 29(7): 1588–1602.
DOI:10.1021/om900917k |
| [101] |
De Bruin T J M, Magna L, Raybaud P, et al. Hemilabile ligand induced selectivity: A DFT study on ethylene trimerization catalyzed by titanium complexes[J]. Organometallics, 2003, 22(17): 3404–3413.
DOI:10.1021/om030255w |
| [102] |
Yang Y, Liu Z, Zhong L, et al. Spin surface crossing betw-een chromium(Ⅰ)/sextet and chromium(Ⅲ)/quartet without deprotonation in SNS-Cr mediated ethylene trimeriz-ation[J]. Organometallics, 2011, 30(19): 5297–5302.
DOI:10.1021/om200722r |
| [103] |
Liu L, Liu Z, Tang S, et al. What triggered the switching from ethylene-selective trimerization into tetramerization over the Cr/(2, 2'-Dipicolylamine) catalysts?[J]. ACS Catal, 2019, 9(11): 10519–10527.
DOI:10.1021/acscatal.9b03340 |
| [104] |
Emrich R, Heinemann O, Jolly P W, et al. The role of meta-llacycles in the Cr-catalyzed trimerization of ethylene[J]. Organometallics, 1997, 16(8): 1511–1513.
DOI:10.1021/om961044c |
| [105] |
Agapie T, Schofer S J, Labinger J A, et al. Mechanistic studies of the ethylene trimerization reaction with chromium-diphosphine catalysts: Experimental evidence for a mechanism involving metallacyclic intermediates[J]. J Am Chem Soc, 2004, 126(5): 1304–1305.
DOI:10.1021/ja038968t |
| [106] |
Overett M J, Blann K, Bollmann A, et al. Mechanistic investigations of the ethylene tetramerization reaction[J]. J Am Chem Soc, 2005, 127(30): 10723–10730.
DOI:10.1021/ja052327b |
| [107] |
McGuinness D S, Suttil J A, Gardiner M G, et al. Ethylene oligomerization with Cr-NHC catalysts: Further insights into the extended metallacycle mechanism of chain growth[J]. Organometallics, 2008, 27(16): 4238–4247.
DOI:10.1021/om800398e |
| [108] |
Chabbra S, Smith D M, Bell N L, et al. First experimental evidence for a bis-ethene chromium(Ⅰ) complex forming from an activated ethene oligomerization catalyst[J]. Sci Adv, 2020, 6(51): 1–6.
|
| [109] |
Agapie T, Labinger J A, Bercaw J E. Mechanistic stud-ies of olefin and alkyne trimerization with chromium catalysts: Deuterium labeling and studies of regiochem-istry using a model chromacyclopentane complex[J]. J Am Chem Soc, 2007, 129(46): 14281–14295.
DOI:10.1021/ja073493h |
| [110] |
Hirscher N A, Labinger J A, Agapie T. Isotopic labeling in ethylene oligomerization: Addressing the issue of 1-octenevs. 1-hexene selectivity[J]. Dalton Trans, 2019, 48(1): 40–44.
DOI:10.1039/C8DT04509G |
| [111] |
Rucklidge A J, McGuinness D S, Tooze R P, et al. Ethylenetetramerization with cationic chromium(Ⅰ) complexes[J]. Organometallics, 2007, 26(10): 2782–2787.
DOI:10.1021/om0701975 |
| [112] |
Monillas W H, Young J F, Yap G P, et al. A well-defin-ed model system for the chromium-catalyzed selective oligomerization of ethylene[J]. Dalton Trans, 2013, 42(25): 9198–9210.
DOI:10.1039/C3DT00109A |
| [113] |
Wang Z, Liu L, Ma X, et al. Effect of an additional donor on decene formation in ethylene oligomerization catalyzed by a Cr/PCCP system: A combined experimental and DFT study[J]. Catal Sci Technol, 2021, 11(13): 4596–4604.
DOI:10.1039/D1CY00423A |
| [114] |
Liu L, Liu Z, Cheng R, et al. Ligand-induced product switching between 4-Methyl-1-pentene and 2-Methyl-1-pentene in Bis(imino)pyridine/V(Ⅲ)-catalyzed propylene dimerization: Cossee-Arlman versus metallacycle mechanism[J]. Organometallics, 2021, 40(11): 1682–1691.
DOI:10.1021/acs.organomet.1c00167 |
| [115] |
Hirscher N A, Arnett C H, Oyala P H, et al. Characteri-zation of Cr-hydrocarbyl species via pulse EPR in thestudy of ethylene tetramerization catalysis[J]. Organo-metallics, 2020, 39(24): 4420–4429.
DOI:10.1021/acs.organomet.0c00521 |
| [116] |
Venderbosch B, Wolzak L A, Oudsen J P H, et al. Role of the ligand and activator in selective Cr-PNP ethene tri- and tetramerization catalysts - a spectroscopic study[J]. Catal Sci Technol, 2020, 10(18): 6212–6222.
DOI:10.1039/D0CY01168A |
| [117] |
Rensburg W J v, Grové C, Steynberg J P, et al. A DFT study toward the mechanism of chromium-catalyzed ethylenetrimerization[J]. Organometallics, 2004, 23(6): 1207–1222.
DOI:10.1021/om0306269 |
| [118] |
Bhaduri S, Mukhopadhyay S, Kulkarni S A. Density functional studies on chromium catalyzed ethylene trimerization[J]. J Org Chem, 2009, 694(9/10): 1297–1307.
|
| [119] |
Do L H, Labinger J A, Bercaw J E. Mechanistic studies of ethylene and α-olefin co-oligomerization catalyzed by chromium-PNP complexes[J]. Organometallics, 2012, 31(14): 5143–5149.
DOI:10.1021/om300492r |
| [120] |
Zilbershtein T M, Kardash V A, Suvorova V V, et al. Deceneformation in ethylene trimerization reaction catalyzed by Cr-pyrrole system[J]. Appl Catal, A: Gen, 2014, 475: 371-378.
|
| [121] |
Wilke G. Organo transition metal compounds as interme-diates in homogeneous catalytic reactions[J]. Pure Appl Chem, 1978, 50: 677–690.
DOI:10.1351/pac197850080677 |
| [122] |
Peitz S, Aluri B R, Peulecke N, et al. An alternative mecha-nistic concept for homogeneous selective ethylene oligomerization of chromium-based catalysts: Binuclear metallacycles as a reason for 1-octene selectivity?[J]. Chem-Eur J, 2010, 16(26): 7670–7676.
DOI:10.1002/chem.201000750 |
| [123] |
McGuinness D S. Olefin oligomerization via metallacycles: Dimerization, trimerization, tetramerization and beyond[J]. Chem Rev, 2011, 111(3): 2321–2341.
DOI:10.1021/cr100217q |
| [124] |
Sydora O L. Selective ethylene oligomerization[J]. Organometallics, 2019, 38(5): 997–1010.
DOI:10.1021/acs.organomet.8b00799 |