 2022, Vol. 36
2022, Vol. 36



过渡金属配合物催化羰基化反应在各种有机化合物的合成中起着重要作用, 因此得到了广泛的研究[1-6]. 通过羰基化反应合成的有机化合物种类很多, 如氢甲酰化[7]合成醛、氨羰基化[8]合成酰胺、氢氨甲基化[9]合成胺、烷氧基羰基化[10]合成酯、氢羰基化[11]合成酸等. 羰基化反应的优势是利用一氧化碳、二氧化碳、甲烷和甲醇等C1单元直接引入羰基, 在合成含羰基的化学品时同时增长碳链[12-13].胺是重要的有机合成原料和化工中间体, 广泛应用于医药、农药、材料和精细化工等领域[14-16]. 传统合成胺的方法包括醇或烷基卤化物的亲核取代和胺的烷基化、醛的还原氨基化、腈的还原等[17-21], 但这些方法通常会产生很多副产物, 原子效率低. 然而, 氢氨甲基化反应作为一种特殊的羰基化反应, 可通过一锅法将廉价易得的胺、烯烃和合成气制备得到有价值的胺[22-23], 为合成胺提供了一种简单高效的合成方法[24-28].
自20世纪50年代初Reppe等[29]报道了第一例HAM以来, 由于其高原子效率和环境友好性, HAM一直受到广泛关注. 他们以[Fe(CO)5]为催化剂, 在苛刻的条件下, 仅得到少量的胺产物. 之后, HAM得到了持续和长足的发展. Rische等[30]将铑催化剂引入到HAM中, 在相对温和的条件下获得了优异的化学选择性和区域选择性, 分别达到96%和89%. Wang等[31]利用RhCl(CO)(TPPTS)2在水-有机两相体系中研究了长链烯烃与二甲胺的化学反应, 在相对温和的条件下(130 ℃, 3 MPa)得到了叔胺的高反应活性和选择性. Fuchs等[32]报道了环二烯与二胺的双酰氨基甲基化反应, 收率高达99%. 本课题组[33]也系统研究了环状烯烃与芳香胺的HAM, 报道了2,5-二氢呋喃与芳香胺通过HAM合成n-芳基胺的策略, 收率达99%. 尽管HAM已经得到了大量的研究[34-39], 但仍然面临许多挑战, 包括更便宜和高效的催化反应体系的开发, 催化剂的回收和复杂底物的扩展等.
4-氨基苯酚作为一种特殊胺类, 是重要的有机合成原料. 由于它既有氨基又有酚羟基官能团, 因此可能表现出不同的反应活性. Xu等[40]报道了氨基酚与碘代芳烃的化学选择性羰基化反应. 他们发现当使用DPPI (1,3-双(二苯基膦基)丙烷)作为配体, K2CO3作为添加剂时, 酚羟基官能团被激活, 以85%的收率得到烷氧基羰基化产物; 当以DIBPP (1,3-双(二异丁基膦基)) 为配体, DBU为添加剂时, 氨基官能团被激活, 以81%收率得到了氨基羰基化产物. 随后, 他们报道了氨基酚的高区域选择性氨羰基化反应. 当以三(4-甲氧基苯基) 膦为配体时, 收率为81% (l∶b = 7.9∶1); 当以1,3,5,7-四甲基-2,4,8-三氧基-6-苯基-6-磷金刚烷为配体时, 收率达96% (l∶b =1∶40). 虽然, 在文献中已经有很多关于氨基酚作为底物的报道, 但氨基酚的HAM还需要深入研究[41-45].
我们以苯乙烯和4-氨基苯酚为模型底物, 通过对溶剂、配体、催化剂前体、添加剂、反应温度、合成气比和总压等因素的筛选, 开发了一种高效、高选择性的催化体系. 结果表明, 添加剂对反应有重要影响. 如图式 1所示, 当以酸(CH3COOH) 为添加剂时, 得到氢氨甲基化产物(1a); 当使用碱(DBU或K2CO3)作为添加剂时, 得到苯乙酮产物(2a).

|
图式 1 苯乙烯与4-氨基苯酚的氢氨甲基化反应 Scheme 1 The hydroaminomethylation (HAM) of styrene with 4-aminophenol |
在本实验中, PPh3通过EtOH再结晶纯化. 其他试剂和溶剂购买后直接使用没有进一步纯化. 使用CDCl3作为溶剂, TMS作为内参, 在Bruker Avance III (400 MHz) 核磁仪上分析产物的1H和13C NMR谱. 通过Agilent Technologies 7890A GC/5975 Ms (EI)和HP-5MS色谱柱(0.25 mm × 30 m, 薄膜: 0.25 μm) 分析产物的GC-Ms谱. 柱层析在粒径0.050 mm硅胶上进行, 薄层色谱分析在硅胶GF254板上进行.
1.2 合成方法将苯乙烯(1 mmol), 4-氨基苯酚(1 mmol), Rh前体(0.005 mmol), 配体(0.025 mmol, P∶Rh = 5∶1), 添加剂和溶剂(5 mL)加入到30 mL的高压釜中. 密封后, 用N2吹扫3次, 加入3 MPa H2和1 MPa CO, 得到的混合物在100 ℃下加热20 h. 然后从油浴中取出, 冷却至室温, 并释放多余的CO和H2. 之后, 反应混合物用乙酸乙酯稀释, 并通过短硅胶柱过滤. 有机溶液经旋转蒸发器浓缩, 以石油醚和乙酸乙酯(5∶1 ~1∶1)的混合洗脱液在硅胶柱上进行柱层析纯化, 得到目标产物. 特别是柱层析过程中未发现苯乙烯残留, 因此有理由相信转化率为99%. 当以碱为添加剂时, 得到了苯乙酮产物(2a). 产物核磁数据如下:
4-[(2-苯丙基)氨基]苯酚(1a):
产率: 186.1 mg (收率: 82%); 棕色油状液体; Rf=0.67 (PE∶EA = 1∶1).
1H NMR (400 MHz, CDCl3) δ 7.30 (t, J =8.0 Hz, 1H), 7.24 - 7.12 (m, 1H), 6.53 (dd, J =8.0, 4.0 Hz, 1H), 4.69 (s, 1H), 3.09 (ddd, J = 4.0, 8.0, 12.0 Hz, 1H), 1.28 (d, J =8.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 148.6, 144.5, 141.5, 128.8, 127.4, 126.7, 116.4, 115.6, 52.6, 39.1, 19.9.
HRMS (ESI): m/z [m+H]+ calcd for C15H18NO 228.138 3, found 228.137 8.
苯乙酮(2a):
产率: 110.3 mg (收率: 92%); 淡黄色油状液体; Rf = 0.67 (PE∶EA = 3∶1).
1H NMR (400 MHz, CDCl3) δ 7.93 - 7.77 (m, 2H), 7.48 (t, J = 8.0 Hz, 1H), 7.38 (t, J = 8.0 Hz, 2H), 2.52 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 197.2, 136.1, 132.1, 127.5, 127.3, 25.6.
GC-Ms: calcd m/z 120.06, found m/z 120.10.
2 结果与讨论 2.1 溶剂、配体和铑前体的影响为了获得苯乙烯与4-氨基苯酚的高催化活性体系, 筛选了各种反应条件, 包括不同溶剂、配体和铑前体的影响, 结果如表 1所示. 首先, 以PPh3(L1)为配体, Rh(CO)2(acac)为催化剂前体(表 1, entries 1-5), 研究了几种常见溶剂(MePh、MeOH、MeCN、THF和EtOH). 结果表明, 当使用MePh作为溶剂时, 没有得到氢氨甲基化的产物(1a), 而苯乙酮(2a)的产率为30%. 由于烯烃在氧化条件下可以通过Wacker氧化生成酮[46], 但该反应在还原条件下得到酮. 当以THF和MeCN为溶剂时, 只得到(2a)产物, 苯乙酮的收率分别为83%和67%. 之后, 研究了不同的醇作为溶剂. 当以MeOH为溶剂时, 氢氨甲基化产物1a的收率为43%, 未得到产物(2a). 接下来, 以EtOH、n-PrOH、i-PrOH和t-BuOH为溶剂时, (1a)收率从35%下降到5%, 仍未得到产物(2a)(表 1, entries 5-8). 结果表明, 当以醇为溶剂时, 没有苯乙酮(2a)产物, 且随着烷基碳原子数的增加, (1a)的收率逐渐降低.因此选择甲醇作为溶剂筛选其他反应条件.
| 表 1 溶剂、配体和铑前体的影响a Table 1 The effects of solvent, ligand and rhodium precursora |
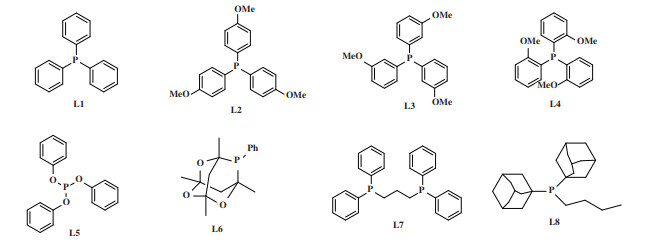
接下来, 以甲醇为溶剂测试不同配体(L1-L8)的影响, 结果如表 1 (entries 9-15) 所示. 当以L2 (三(4-甲氧基苯基) 膦) 为配体时, 氢氨甲基化产物(1a)的收率为55%, 未得到产物苯乙酮(2a). 以L3 (三(3-甲氧基苯基) 膦) 为配体时, (1a)的收率最高, 达到66%, 未得到产物(2a). 当使用L4 (三(2-甲氧基苯基) 膦) 作为配体时, 总收率下降到66%. 亚磷酸配体也广泛应用于羰基化反应中, 所以当使用L5 (三苯基亚磷酸) 作为配体时, (1a)的收率为60%, 没有得到产物(2a). 之后, 使用空间位阻较大的配体L6 (1,3,5,7-四甲基-2,4,8-三氧基-6-苯基-6-磷) 和L7 (1,3-双(二苯基磷)丙烷), 得到(1a)的产率分别为21%和34%, (2a)的产率分别为17%和21%, 表明空间位阻较大的配体对反应有不利影响. 特别是, 当使用L8 (丁基二-1-金刚酰基膦) 作为配体时, 没有得到任何产物. 对比以上实验结果可以看出, 当使用L3时, 反应的选择性和总收率最高. 因此, 选择L3作为配体进行其他条件的筛选.
为了提高该反应的选择性和反应性, 以MeOH为溶剂, L3为配体, 研究了几种不同的催化剂前体(表 1, entries 16-20). 结果表明, 当采用Rh(cod)2BF4、[Rh(cod)]Cl2、[Cp*RhCl2]2、RhCl3·3H2O等不同的催化剂前体时, 氢氨甲基化产物(1a)的收率大致相等, 没有得到苯乙酮产物(2a). 然而, 当使用RhCl-(PPh3)3作为催化剂前体时, 产物(1a)的收率提高到70%, 未得到产物(2a), 表明该反应获得了高选择性和高反应活性的氢氨甲基化产物. 由于RhCl(PPh3)3中存在配体PPh3, 因此推测催化剂前体中的配体可能对选择性和反应性有积极影响. 当不加入L3时, (1a)的收率下降到10%, (2a)的收率为62%.因此, 可能是由于RhCl(PPh3)3和L3中的配体PPh3的协同作用导致了该反应具有较高的反应活性和选择性. 因此, 进一步使用RhCl(PPh3)3作为催化剂前体.
2.2 添加剂及其含量的影响添加剂在有机合成反应中起着至关重要的作用. 因此, 对包括酸和碱在内的几种不同添加剂的效果进行了测试, 如表 2所示. 结果表明, 当使用酸作为添加剂时, 产物(2a)的收率为0(表 2, entries 1-8), 表明在酸性条件下只发生了氢氨甲基化反应, 其中不同酸性添加剂的pH顺序为: P-TSA < 5-ClSA < C6H5COOH < CH3COOH < C2H5COOH < C3H7COOH < H3BO3. 首先, 当使用酸性较低的H3BO3作为添加剂时, 产物(1a)的收率为30%, 反应活性降低. 当使用C3H7COOH、C2H5COOH和CH3COOH作为添加剂时, 可以看到, 随着添加剂酸度的增加, 产物(1a)的收率从33%增加到82%. 特别是当以CH3COOH为添加剂时, 产物(1a)的收率最高. 之后继续增加添加剂的酸度, 例如使用C6H5COOH、5-ClSA和P-TSA作为添加剂, (1a)的收率从82%下降到13%. 此外还尝试使用5% H3BO3和10% 5-ClSA等混合酸作为添加剂, 结果表明(1a)的收率为68%, 总收率下降. 相应地也考察了两种不同类型碱的影响(表 2, entries 9-10). 结果表明, 当使用碱作为添加剂时, 只得到苯乙酮(2a), 表明在碱存在下不会发生氢氨甲基化反应. 可以看出, 当以弱碱性的有机碱DBU作为添加剂时, (2a)的收率高达92%, 而当以碱性较强的无机碱K2CO3作为添加剂时, (2a)的收率降低到63%, 这些结果表明, 添加剂的碱性影响了反应的活性.
| 表 2 添加剂及其含量的影响a Table 2 The effects of additives and the amount of additivea |
以上实验结果表明, 该催化体系存在一个最佳的pH值范围, 从而导致反应在氢氨甲基化或获得苯乙酮的反应间有选择性. 因此, 以CH3COOH作为添加剂进一步考察了添加剂用量对反应的影响, 如表 2 (entries 11-14) 所示. 结果表明, 当添加剂CH3COOH的添加量减少到10%(摩尔分数)时, 产物(1a)的收率下降到41%.同样, 当CH3COOH的添加量提高到30%(摩尔分数)时, 产物(1a)的收率下降到44%. 表明增加或减少添加剂的用量都会降低反应的反应活性. 因此, 为了保证反应的高选择性和高反应性, 确定了最佳的添加剂用量为20%(摩尔分数).
2.3 温度和压力的影响进一步考察了温度和压力对反应的影响, 结果如表 3所示. 首先, 当温度为60 ℃时, 未得到氢氨甲基化反应产物(1a), 苯乙酮(2a)的收率为25%. 当温度提高到80 ℃时, 仍未得到产物(1a), 苯乙酮(2a)的收率提高到51%. 当温度继续提高到120 ℃时, (1a)的收率较100 ℃时下降到38%, 未得到(2a)产物. 这说明低温条件下不利于氢氨基甲基化产物(1a)的形成, 温度过低或过高都会降低反应的活性, 所以最优反应温度是100 ℃. 最后, 考察了合成气压力的影响. 当总压强为4 MPa且H2∶CO的比率为2∶2时, 产物(1a)的收率为53%. 改变H2∶CO的比率为1∶3时, 产物(1a)的收率下降至38%. 接下来, 当总压强下降至3 MPa且H2∶CO的比率是1.5∶1.5时, 产物(1a)的收率为41%. 可以看出, 当氢气含量较高时导致产生更多的氢氨甲基化产物(1a). 由此可知, 最佳反应压力为4 MPa, 且H2∶CO比为3∶1.
| 表 3 温度和压力的影响a Table 3 The effects of temperature and pressurea |
基于以上实验结果并结合文献报道[9], 提出了苯乙烯与4-氨基苯酚的氢氨甲基化反应可能的机理, 如图示2所示. 由于4-氨基苯酚具有胺基和羟基官能团的特性, 有两种可能的途径. 首先, 铑前体、配体和合成气形成的配合物A催化苯乙烯反应生成中间产物B, 然后中间产物B经过氢甲酰化反应生成产物C. 之后, 中间体C与4-氨基苯酚缩合得到中间产物D. 然后, 当以CH3COOH为添加剂时, 通过路径I反应得到氢氨甲基化产物(1a), 中间产物D脱水生成亚胺产物E, 之后亚胺E被氢还原生成产物(1a), 活性催化剂进入下一个催化反应循环. 当碱作为添加剂时, 反应遵循路径II, 由于溶剂中存在水和操作过程中微量的氧, 中间产物G形成, 且利用有机碱DBU可获得更多的苯乙酮产物. GC-Ms分析证实, 用苯胺取代4-氨基酚得到的酰胺和苯乙酮相似. 在还原气氛下合成苯乙酮产品无疑将为酮类有机化合物及其中间体的合成提供新的思路和方法.

|
图式 2 苯乙烯与4-氨基苯酚氢化氨甲基化的可能机理 Scheme 2 Possible mechanism of hydroaminomethylation of styrene with 4-aminophenol |
综上所述, 我们建立了一个高效的催化体系, 在羰基化反应中实现了苯乙烯与4-氨基苯酚的高反应性和高选择性调控. 通过考察溶剂、催化剂前体、配体、添加剂及合成气比例对反应的影响, 确定了最优反应条件. 当使用酸作为添加剂时, 主要发生氢氨甲基化反应生成胺, 产物收率最高可达82%. 然而, 当使用碱作为添加剂时, 该反应主要生成苯乙酮, 收率为92%, 这与氧化条件下烯烃发生Wacker氧化反应生成酮不同, 在还原条件下也得到了苯乙酮. 最后, 预测了苯乙烯与4-氨基苯酚可能的反应机理, 说明了两种不同的反应途径.
| [1] |
a. Selander N, Szabo K. Catalysis by palladium pincer comp-lexes[J]. Chem Rev, 2011, 111(3): 2048-2076. b. Yang Shu-qing(杨淑晴), Liu Ye(刘晔). Preparation of acylhydrazines via carbonylation and their applications as intelligent materials(酰肼类化合物的羰基化制备及其在智能材料合成中的应用)[J]. J Mol Catal (China)(分子催化), 2021, 35(5): 456-470. |
| [2] |
a. Enthale S, Company A. Palladium-catalysed hydroxy-lation and alkoxylation[J]. Chem Soc Rev, 2011, 40(10): 4912-4924. b. Sun Ze-ping(孙泽平), Wu Jian-bing(武建兵), Li Peng(李鹏), et al. Effect of citric acid modification of ZSM-5 zeolite on vapor-phase dimethoxymethane carbonylation(柠檬酸处理对ZSM-5分子筛甲缩醛气相羰基化性能的影响)[J]. J Mol Catal (China)(分子催化), 2021, 35(1): 22-30. c. Cao Yan-wei(曹彦伟), Zhang Xue-hua(张雪华), HeLin(何林). Recent advance in synthesis of urea by oxidative carbonylation of amine(胺氧化羰基化合成脲的研究进展)[J]. J Mol Catal (China)(分子催化), 2020, 34(2): 182-192. |
| [3] |
a. Wu X, Neumann H, Beller M. Synthesis of heterocycles via palladium-catalyzed carbonylations[J]. Chem Rev, 2013, 113(1): 1-35. b. Aisha-Nulahong(艾沙·努拉洪), Fang Ya-ping(方亚平), Gao Xi-ran(高希然), et al. Effect of ZSM-5 zeolites with different grains on carbonylation of syngas(不同晶粒ZSM-5沸石分子筛对合成气羰基化反应性能的影响)[J]. J Mol Catal (China)(分子催化), 2020, 34(2): 105-115. |
| [4] |
Wu X, Neumann H. Ruthenium and rhodium-catalyzed carbonylation reactions[J]. ChemCatChem, 2012, 4(4): 447–458.
DOI:10.1002/cctc.201200069 |
| [5] |
Etayo P, Vidal A. Rhodium-catalysed asymmetric hydrog-enation as a valuable synthetic tool for the preparation of chiral drugs[J]. Chem Soc Rev, 2013, 42(2): 728–754.
DOI:10.1039/C2CS35410A |
| [6] |
Fleischer I, Gehrtz P, Hirschbeck V, et al. Carbonylations of alkenes in the total synthesis of natural compounds[J]. Synthesis, 2016, 48(11): 1573–1596.
DOI:10.1055/s-0035-1560431 |
| [7] |
Brezny A, Landis C. Recent developments in the scope, practicality, and mechanistic understanding of enantio-selective hydroformylation[J]. Acc Chem Res, 2018, 51(9): 2344–2354.
DOI:10.1021/acs.accounts.8b00335 |
| [8] |
Kumar D, Vemula S, Cook G. Recent advances in the catalyticsynthesis of α-ketoamides[J]. ACS Catal, 2016, 6(8): 4920–4945.
DOI:10.1021/acscatal.6b01116 |
| [9] |
Chen C, Dong X Q, Zhang X. Recent progress in rhodium-catalyzed hydroaminomethylation[J]. Org Chem Front, 2016, 3(10): 1359–1370.
DOI:10.1039/C6QO00233A |
| [10] |
Kalck P, Urrutigoïty M. Recent improvements in the alkoxycarbonylation reaction catalyzed by transition metal complexes[J]. Inorg Chim Acta, 2015, 431: 110–121.
DOI:10.1016/j.ica.2015.02.007 |
| [11] |
Gaydou M, Moragas T, Julia-Hernandez F, et al. Site-selective catalytic carboxylation of unsaturated hydrocarbons with CO2 and water[J]. J Am Chem Soc, 2017, 139(35): 12161–12164.
DOI:10.1021/jacs.7b07637 |
| [12] |
Sumino S, Fusano A, Fukuyama T, et al. Carbonylation reactions of alkyl iodides through the interplay of carbon radicals and Pd catalysts[J]. Acc Chem Res, 2014, 47(5): 1563–1574.
DOI:10.1021/ar500035q |
| [13] |
Li W, Liu C, Zhang H, et al. Palladium-catalyzed oxidative carbonylation of N-allylamines for the synthesis of beta-lactams[J]. Angew Chem Int Ed, 2014, 53(9): 2443–2446.
DOI:10.1002/anie.201309081 |
| [14] |
Yamamoto Y, Radhakrishnan U. Palladium catalysed pronucleophile addition to unactivated carbon-carbon multiple bonds[J]. Chem Soc Rev, 1999, 28(3): 199–207.
DOI:10.1039/a806581k |
| [15] |
Müller T, Beller M. Metal-initiated amination of alkenes and alkynes[J]. Chem Rev, 1998, 98(2): 675–704.
DOI:10.1021/cr960433d |
| [16] |
D'Alessandro D, Smit B, Long J. Carbon dioxide capture: Prospects for new materials[J]. Angew Chem Int Ed, 2010, 49(35): 6058–6082.
DOI:10.1002/anie.201000431 |
| [17] |
Clerici A, Cannella R, Panzeri W, et al. TiCl3/PhN2+-mediatedradical addition of ethers to aldimines generated in situ under aqueous conditions[J]. Tetrahedron Lett, 2005, 46(48): 8351–8354.
DOI:10.1016/j.tetlet.2005.09.158 |
| [18] |
Angelovski G, Eilbracht P. Synthesis of hydroquinone-, biphenol-, and binaphthol-containing aza macroheterocycles via regioselective hydroformylation and reductiveamination[J]. Tetrahedron, 2003, 59(41): 8265–8274.
DOI:10.1016/j.tet.2003.08.012 |
| [19] |
Quach T, Batey R. Ligand- and base-free copper(Ⅱ)-catalyzed C-N bond formation: Cross-coupling reactions of organoboron compounds with aliphatic amines and anilines[J]. Org Lett, 2003, 5(23): 4397–4400.
DOI:10.1021/ol035681s |
| [20] |
Gurak J, Tran V, Sroda M, et al. N-alkylation of 2-pyri-done derivatives via palladium(Ⅱ)-catalyzed directed alkene hydroamination[J]. Tetrahedron, 2017, 73(26): 3636–3642.
DOI:10.1016/j.tet.2017.03.091 |
| [21] |
Amer M, Aziz M, Sheha , et al. Recent advances in chemistry and pharmacological aspects of 2-pyridone scaffolds[J]. J Saudi Chem Soc, 2021, 25: 101259–101304.
DOI:10.1016/j.jscs.2021.101259 |
| [22] |
Behr A, Levikov D, Nürenberg E. Rhodium catalyzed one-step hydroamidation of cyclopentadiene and dicyclop-entadiene[J]. Catal Sci Technol, 2015, 5(5): 2783–2787.
DOI:10.1039/C5CY00168D |
| [23] |
Cousin K, Vanbésien T, Monflier E, et al. One pot synth-esis of aminohydroxylated triglycerides under aqueous biphasic conditions[J]. Cata Commun, 2019, 125: 37–42.
DOI:10.1016/j.catcom.2019.03.018 |
| [24] |
Behr A, Levikov D, Nürenberg E. Rhodium-catalyzed hydroaminomethylation of cyclopentadiene[J]. RSC Adv, 2015, 5(75): 60667–60673.
DOI:10.1039/C5RA10870E |
| [25] |
Oliveira D, Gutiérrez M, Villarreal J, et al. Sustainable route to biomass-based amines: Rhodium catalyzed hydroa-minomethylation in green solvents[J]. Appl Cata A, 2019, 574: 97–104.
DOI:10.1016/j.apcata.2019.02.003 |
| [26] |
Briggs J, Klosin J, Whiteker G. Synthesis of biologically active amines via rhodium-bisphosphite-catalyzed hydro-aminomethylation[J]. Org Lett, 2005, 7(22): 4795–4798.
DOI:10.1021/ol050848y |
| [27] |
Ahmed M, Seayad A, Jackstell R, et al. Amines made easily: A highly selective hydroaminomethylation of olefins[J]. J Am Chem Soc, 2003, 125(34): 10311–10318.
DOI:10.1021/ja030143w |
| [28] |
Huang L, Arndt M, Goossen K, et al. Late transition metal-catalyzed hydroamination and hydroamidation[J]. Chem Rev, 2015, 115(7): 2596–2697.
DOI:10.1021/cr300389u |
| [29] |
Reppe W, Vetter H. Synthesen mit metallcarbonylwas-serstoffe[J]. Justus Liebigs Ann Chem, 1953, 582: 133–139.
DOI:10.1002/jlac.19535820107 |
| [30] |
Rische T, Kitsos B, Eilbracht P. Selective one-potsynthesis of symmetrically and unsymmetrically substitutedamines via rhodium catalysed multiple alkylations of ammonia or primary amines under hydroformylation conditions[J]. Tetrahedron, 1998, 54: 2723–2742.
DOI:10.1016/S0040-4020(98)83008-1 |
| [31] |
Wang Y, Luo M, Li Y, et al. The catalytic hydroam-inomethylation of long chain alkenes with dimethylamine in aqueous-organic two-phase system[J]. Appl Catal A, 2004, 272(1/2): 151–155.
|
| [32] |
Fuchs S, Steffen M, Dobrowolski A, et al. Secondary diamines as a monomer from bis-hydroaminomethylation of industrial cyclic dienes[J]. Catal Sci Technol, 2017, 7(21): 5120–5127.
DOI:10.1039/C7CY01050H |
| [33] |
Zheng Z, Wang L. One-Pot synthesis of N-arylated aminesby hydroaminomethylation of 2, 5-dihydrofuran with aromatic amines[J]. Synthesis, 2019, 51(7): 1585–1594.
DOI:10.1055/s-0037-1610681 |
| [34] |
Khan S, Bhanage B. Selective hydroaminomethylation of olefins using simple and efficient Rh-phosphinite complex catalyst[J]. Appl Organomet Chem, 2013, 27(12): 711–715.
DOI:10.1002/aoc.3046 |
| [35] |
Seidensticker T, Vosberg J, Ostrowski K, et al. Rhodium-catalyzed bis-hydroaminomethylation of linear aliphatic alkenes with piperazine[J]. Adv Synth Catal, 2016, 358(4): 610–621.
DOI:10.1002/adsc.201500896 |
| [36] |
Fuchs S, Rosler T, Grabe B, et al. Synthesis of primary amines via linkage of hydroaminomethylation of olefins and splitting of secondary amines[J]. Appl Catal A, 2018, 550: 198–205.
DOI:10.1016/j.apcata.2017.11.010 |
| [37] |
Oliveira K, Carvalho S, Duarte M, et al. Phospholes asefficient ancillaries for the rhodium-catalyzed hydroform-ylation and hydroaminomethylation of estragole[J]. Appl Catal A, 2015, 497: 10–16.
DOI:10.1016/j.apcata.2015.02.028 |
| [38] |
Crozet D, Kefalidis C, Urrutigoïty M, et al. Hydroamin-omethylation of styrene catalyzed by rhodium complexes containing chiral diphosphine ligands and mechanistic studies: Why is there a lack of asymmetric induction?[J]. ACS Catal, 2014, 4(2): 435–447.
DOI:10.1021/cs400906b |
| [39] |
Zheng X, Xu K, Zhang X. Highly selective bisphosphine ligands for asymmetric hydroformylation of heterocyclic olefins[J]. Tetrahedron Lett, 2015, 56(9): 1149–1152.
DOI:10.1016/j.tetlet.2015.01.052 |
| [40] |
Xu T, Alper H. Pd-catalyzed chemoselective carbonylation of aminophenols with iodoarenes: Alkoxycarbonylation vs aminocarbonylation[J]. J Am Chem Soc, 2014, 136(49): 16970–16973.
DOI:10.1021/ja508588b |
| [41] |
Xu T, Sha F, Alper H. Highly ligand-controlled regiose-lective Pd-catalyzed aminocarbonylation of styrenes with aminophenols[J]. J Am Chem Soc, 2016, 138(20): 6629–6635.
DOI:10.1021/jacs.6b03161 |
| [42] |
Sharma M, Sarmah B, Bhattacharya P, et al. Dicarbon-ylrhodium(I) complexes of aminophenols and their catalytic carbonylation reaction[J]. Appl Organomet Chem, 2007, 21(4): 255–263.
DOI:10.1002/aoc.1193 |
| [43] |
Noujima A, Mitsudome T, Mizugaki T, et al. Gold nanoparticle-catalyzed cyclocarbonylation of 2-aminoph-enols[J]. Green Chem, 2013, 15(3): 608–611.
DOI:10.1039/c2gc36851j |
| [44] |
Liu B, Yin M, Gao H, et al. Synthesis of 2-aminoben-zoxazoles and 3-aminobenzoxazines via palladium-catalyzedaerobic oxidation of o-aminophenols with isocyanides[J]. J Org Chem, 2013, 78(7): 3009–3020.
DOI:10.1021/jo400002f |
| [45] |
Neumann K, Lindhardt A, Andersen B, et al. Access to 2-(het)aryl and 2-styryl benzoxazoles via palladium-catalyzed aminocarbonylation of aryl and vinyl bromides[J]. Org Lett, 2015, 17(9): 2094–2097.
DOI:10.1021/acs.orglett.5b00642 |
| [46] |
Zhang Z, Kumamoto Y, Hashiguchi T, et al. Wacker oxidation of terminal alkenes over ZrO2-supported Pd nanoparticles under acid- and cocatalyst-free conditions[J]. ChemSusChem, 2017, 10(17): 3482–3489.
DOI:10.1002/cssc.201701016 |