 2023, Vol. 37
2023, Vol. 37



2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049, China
二氧化碳(CO2)是一种主要的温室气体, 同时也是一种价廉丰富、无毒且可再生的C1资源, 将其固定转化为增值化学品是缓解全球温室效应问题的一种有效方式[1−3]. 由于CO2的热力学稳定性和动力学惰性[4], 因此, 需要借助有效的催化剂来降低CO2的反应活化能. 迄今为止, 已经开发了多种方法将CO2转化为不同的化学品[2]. 其中, CO2与环氧化合物(PO)反应合成环状碳酸酯具有100%原子经济性, 是CO2合成高附加值化学品中研究的热点. 环状碳酸酯被广泛应用于极性非质子溶剂、锂离子电池的电解质、聚合物材料的前体、燃料添加剂和精细化学品合成的中间体[5]. 目前, 用于CO2与PO反应合成环状碳酸酯的催化剂包括有机催化剂和金属基催化剂[6−8]. 这些催化体系需要路易斯酸性/碱性位点来活化环氧化物/CO2, 并需要高能亲核试剂来打开PO环. 已经报道了多种均相二元催化体系, 其涉及金属配合物和助催化剂的组合[9−12]. 二元催化剂需要亲核助催化剂, 造成额外的浪费和环境污染, 同时, 自由接近亲核部分的概率也很低. 为了克服这一缺点, 开发了在没有助催化剂和温和的条件下促进环状碳酸酯形成的单组份双功能金属催化剂体系. 比如通过共价连接到路易酸金属中心的季铵盐、季膦盐和离子液体盐作为亲核试剂[13−15], 路易斯酸金属中心携带有机碱作为亲核试剂的配体[16−17], 路易斯酸金属中心携带不稳定的轴向卤化物可逆解离产生亲核的卤化物阴离子[18]以及路易酸金属中心和亲核抗衡离子包含在离子对的同一分子内等[19−20]. 这些催化剂表现出比二元催化剂更高的催化活性, 因为路易斯酸金属中心和亲核阴离子分子内的协同作用增强了催化活性. 然而, 在催化剂设计中仍然需要进一步开发具有简单配体骨架的新型稳定的双功能催化剂.
配体结构和性质对金属配合物具有至关重要的影响, 氮杂卡宾(NHC)配体因其独特的物理化学性质而被广泛研究[21−22]. 由于NHC-金属键非常强能够防止结构分解, N-杂环卡宾及其金属配合物被用于多种有机转化[23−25]. 锌盐是具有强路易斯酸性的廉价、低毒和稳定的金属盐, 并且锌基配合物已被证明是用于CO2与环氧化物偶联的有效催化剂[26−29]. 目前, 氮杂卡宾锌络合物作为单组分双功能催化剂用于CO2转化为环状碳酸酯的研究报道很少. 基于这些, 我们设计合成了一系列新型芘标记含溴亲核阴离子的氮杂环卡宾前驱体化合物, 活化成游离态的氮杂环卡宾与卤化锌配位后, 筛选出优异的氮杂卡宾锌配合物作为有效的单组份多功能催化剂, 在无溶剂和助催化剂的条件下对CO2和PO偶联合成环状碳酸酯显示出较高的催化活性. 系统考察了其反应参数包括温度、压力、反应时间以及催化剂量等的影响, 结合FTIR表征等, 推测了反应机理.
1 实验部分 1.1 试剂和仪器除另有说明, 所有试剂均为分析级, 直接使用. 使用Bruker AvanceTM III 400 MHz超导核磁共振谱仪, 将含有TMS的DMSO-D6作为溶剂, 获得1H NMR和13C NMR谱, 化学位移为δ, 耦合常数J的单位是Hz. 使用德国Bruker 400MHz超导核磁共振谱仪获得13C CP-MAS NMR谱. 使用ESCALAB 250Xi(美国)X射线光电子能谱仪进行表面元素组成价态分析. 使用Agilent 725-ES光谱仪进行无机元素定性和定量分析. 使用VERTEX 70v(德国)采集FT-IR谱图.
1.2 合成芘标记的NHC前驱体分子的过程化合物1(Comp. 1): 2-(2-溴乙氧基)芘的制备: 如图1所示, 在100 mL圆底烧瓶中, 分别加入1-羟基芘 (2.18 g, 10 mmol)、Cs2CO3 (6.5 g, 20 mmol)、1,2-二溴乙烷(7.48 g, 40 mmol)和乙腈(35 mL), 在80 ℃下搅拌回流24 h. 冷却后, 加水(50 mL), 用二氯甲烷(3×30 mL)萃取, 萃取液用硫酸镁进行干燥. 最后, 通过柱层析(乙酸乙酯∶石油醚= 1∶4)对所得产物进行纯化, 得到黄色固体(2.3 g, 70%收率).

|
图 1 芘修饰的NHC前驱体化合物的合成 Fig.1 Synthesis of NHC precursor compounds modified with pyrene |
1H NMR (400 MHz, DMSO-d6) δ : 8.44 (d, J = 9.2 Hz, 1H), 8.26 (d, J = 8.5 Hz, 1H), 8.22 (dd, J = 7.6, 4.7 Hz, 2H), 8.17 (d, J = 9.2 Hz, 1H), 8.09 (d, J = 8.9 Hz, 1H), 8.05 (d, J = 7.7 Hz, 1H), 8.03 – 7.98 (m, 1H), 7.79 (d, J = 8.5 Hz, 1H), 4.72 (dd, J = 6.1, 4.5 Hz, 2H), 4.04 (dd, J = 6.1, 4.4 Hz, 2H).
13C NMR (101 MHz, DMSO) δ: 152.34, 131.67, 127.73, 127.00, 126.43, 125.49, 125.42, 125.02, 124.80, 121.29, 120.09, 110.89, 69.33, 32.34.
化合物2: 1-(2-(芘-2-酰氧基)乙基)- 1h -苯并咪唑的制备: 将苯并咪唑(1.18 g, 10 mmol)和氢氧化钾(1.13 g, 12 mmol)溶于乙腈(50 mL)的混合物, 在80 ℃下先回流30 min, 再加入化合物1 (3.26 g, 10 mmol), 继续回流24 h. 冷却至室温后, 向反应溶液中加入20 mL正己烷后过滤, 所得固体用正己烷洗涤3次, 真空干燥得到淡黄色固体(2.7 g, 75%收率).
1H NMR (400 MHz, DMSO-d6) δ: 8.52 (s, 1H), 8.23 – 8.17 (m, 4H), 8.04 (dt, J = 17.1, 8.5 Hz, 4H), 7.86 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 4.94 (dd, J = 11.4, 6.4 Hz, 2H), 4.71 (t, J = 5.0 Hz, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 152.39, 145.19, 134.50, 131.62, 131.45, 127.78, 127.70, 126.94, 126.87, 126.39, 125.40, 125.38, 125.34, 124.96, 124.75, 122.79, 122.03, 121.18, 119.93, 119.68, 111.16, 110.22, 67.76, 44.38.
化合物3: 苯并咪唑盐(3a, 3b)的合成: 向20 mL乙腈中加入化合物2 (2 mmol, 724 mg)和2.2 mmol卤代烃, 在80 ℃下回流24 h. 过滤后固体用正己烷洗涤, 真空干燥后产品为黄色固体物质(3a: 800 mg, 产率75%; 3b: 560 mg, 产率56%).
3a: 1H NMR (400 MHz, DMSO-d6) δ: 10.33 (s, 1H), 8.39 – 8.35 (m, 1H), 8.27 – 8.17 (m, 5H), 8.09 – 7.98 (m, 6H), 7.77 (t, J = 8.0 Hz, 2H), 7.69 (t, J = 7.8 Hz, 1H), 7.53 (dd, J = 6.8, 2.9 Hz, 2H), 7.32 – 7.26 (m, 3H), 5.85 (s, 2H), 5.26 (t, J = 4.8 Hz, 2H), 4.88 (t, J = 4.9 Hz, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 152.01, 143.61, 134.34, 132.00, 131.61, 131.41, 131.30, 129.40, 129.21, 128.80, 127.68, 127.32, 127.03, 126.98, 126.34, 125.56, 125.11, 124.86, 124.42, 121.12, 114.76, 114.47, 110.32, 66.68, 50.41, 47.22.
3b: 1H NMR (400 MHz, DMSO-d6) δ: 10.39 (s, 1H), 8.39 (t, J = 15.2 Hz, 1H), 8.22 (dd, J = 14.5, 8.5 Hz, 4H), 8.14 (d, J = 8.2 Hz, 1H), 8.11–7.94 (m, 4H), 7.85–7.68 (m, 3H), 5.28 (d, J = 4.3 Hz, 2H), 5.02–4.77 (m, 2H), 4.58 (t, J = 7.0 Hz, 2H), 1.97–1.73 (m, 2H), 1.43–1.10 (m, 2H), 0.78 (t, J = 7.3 Hz, 3H).
13C NMR (101 MHz, DMSO) δ: 151.5, 142.9, 131.2, 131.0, 130.8, 127.1, 126.6, 126.4, 125.8, 124.9, 124.7, 124.5, 124.3, 123.9, 120.6, 119.1, 114.1, 113.8, 109.8, 66.3, 46.55, 30.5, 19.0, 13.3.
化合物4: 双苯并咪唑单体的合成: 向30 mL DMSO中加入BIM(17 mmol, 2 g)和KOH(25 mmol, 1.4 g), 先在80 ℃下搅拌90 min, 冷却至室温后, 加入卤代烃8.5 mmol, 搅拌3 h后倒入300 mL冷的去离子水, 过滤除掉DMSO, 去离子水洗涤3次后, 真空干燥后得白色固体(3a: 2.23 g , 产率90%).
1H NMR (400 MHz, DMSO-d6) δ: 8.23 (s, 2H), 7.61 (dd, J = 24.1, 7.8 Hz, 4H), 7.22 (dt, J = 18.8, 7.2 Hz, 4H), 4.28 (d, J = 5.8 Hz, 4H), 1.85 – 1.70 (m, 4H).13C NMR (101 MHz, DMSO) δ 144.42, 143.77, 134.18, 122.73, 121.94, 119.87, 110.87, 43.99, 27.22.
化合物5a: 苯并咪唑盐的合成: 向20 mL乙腈中加入化合物4(4 mmol, 1.16 g)和化合物1(4 mmol, 1.3 g), 在80 ℃下搅拌30 h后, 直接旋蒸至饱和溶液加入正己烷后析出的固体分别用乙腈正己烷洗涤2次后, 真空干燥得淡黄色固体(1.4 g, 产率57%).
1H NMR (400 MHz, DMSO-d6) δ: 10.12 (s, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.19 (d, J = 4.1 Hz, 5H), 8.06 (ddd, J = 10.5, 8.0, 5.3 Hz, 4H), 7.82 – 7.70 (m, 3H), 7.63 – 7.54 (m, 2H), 7.44 – 7.37 (m, 1H), 7.20 – 7.09 (m, 2H), 5.21 (t, J = 4.8 Hz, 2H), 4.85 (t, J = 4.8 Hz, 2H), 4.58 (d, J = 6.6 Hz, 2H), 4.24 – 4.16 (m, 2H), 1.92 – 1.82 (m, 4H).
13C NMR (101 MHz, DMSO-d6) δ: 151.51, 143.93, 143.87, 143.38, 142.88, 131.34, 131.11, 130.98, 127.17, 126.69, 126.65, 126.52, 126.43, 125.84, 125.07, 124.62, 124.35, 122.19, 122.12, 121.39, 114.09, 113.68, 110.17, 109.84, 66.28, 46.57, 43.47, 43.33, 26.72, 26.17.
化合物5b: 苯并咪唑盐的合成: 向25 mL乙腈中加入化合物4(4 mmol, 1.16 g)和化合物1(8.2 mmol, 2.6 g), 在80 ℃下搅拌30 h后, 直接旋蒸至饱和溶液加入正己烷后析出的固体用二氯甲烷洗涤3次后, 真空干燥得乳白色固体(2.36 g, 产率62%).
1H NMR (400 MHz, DMSO-d6) δ: 10.16 (s, 2H), 8.28 (d, J = 8.4 Hz, 2H), 8.20 (s, 1H), 8.18−8.17 (s, 4H), 8.15 (d, J = 2.9 Hz, 2H), 8.13 (s, 1H), 8.03 (s, 2H), 8.01 (s, 1H), 8.00 (d, J = 2.0 Hz, 2H), 7.97 (s, 2H), 7.95 (s, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.73 – 7.66 (m, 4H), 7.55 (t, J = 7.8 Hz, 2H), 5.14 (t, J = 4.8 Hz, 4H), 4.78 (t, J = 4.9 Hz, 4H), 4.56 (d, J = 5.8 Hz, 4H), 2.02 (d, J = 5.1 Hz, 4H).
13C NMR (101 MHz, DMSO) δ: 151.96, 143.33, 131.80, 131.59, 131.32, 127.00, 126.79, 126.32, 125.52, 125.22, 125.11, 124.76, 124.70, 124.36, 121.14, 119.55, 114.54, 113.99, 110.25, 66.85, 46.98, 46.61, 25.87.
1.3 卤化锌在NHC上的固定在惰性气氛下, 向Schlenk管中先引入化合物3b(0.8 mmol, 400 mg)、叔丁醇钾(1.2 mmol, 136 mg)和溴化锌(0.8 mmol, 180 mg), 加入20 mL超干的THF, 在50 ℃下搅拌24 h后, 冷却后加入正己烷(20 mL), 得到白色固体, 用正己烷(3×30 mL)进行洗涤, 后干燥过夜得到化合物NHC-3b-ZnBr2.
1H NMR (400 MHz, DMSO-d6) δ: 8.08 – 7.93 (m, 5H), 7.91 (d, J = 7.6 Hz, 1H), 7.84 – 7.69 (m, 3H), 7.61 (dd, J = 16.9, 8.3 Hz, 2H), 7.46 (d, J = 7.7 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 5.11 (t, J = 5.1 Hz, 2H), 4.84 (t, J = 5.1 Hz, 2H), 4.27 (t, J = 7.4 Hz, 2H), 1.85 – 1.70 (m, 2H), 1.23 (d, J = 7.8 Hz, 3H), 0.74 (t, J = 7.3 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ: 169.54, 163.87, 153.02, 146.59, 131.67, 131.11, 129.05, 127.77, 126.92, 126.85, 126.49, 126.44, 126.38, 125.42, 125.21, 124.94, 124.70, 124.58, 121.32, 120.19, 119.28, 116.77, 111.86, 111.06, 109.72, 50.30, 25.29, 23.34.
在惰性气氛下, 向Schlenk管中先加入化合物5a(2.6 mmol, 1.6 g)和叔丁醇钾(5.2 mmol, 582 mg), 加入50 mL超干THF后, 在40 ℃下搅拌10 h. 冷却至室温后, 加去离子水(50 mL), 用二氯甲烷(3×30 mL)萃取, 用无水碳酸钠干燥后通过真空旋转蒸发浓缩至饱和溶液, 向其中加入正己烷(30 mL), 得到黄色固体, 用正己烷(3×30 mL)进行洗涤, 后干燥过夜得到游离卡宾化合物6(NHC-PDBI). 由于对应于咪唑环的酸性NCHN信号的消失, 通过1H NMR分析清楚地证实了游离卡宾的形成.
1H NMR (400 MHz, DMSO-d6) δ: 8.22 – 8.12 (m, 5H), 8.06 – 7.92 (m, 7H), 7.62 (d, J = 3.3 Hz, 4H), 7.21 (dd, J = 16.8, 7.6 Hz, 5H), 6.63 – 6.50 (m, 1H), 4.45 (dt, J = 16.5, 5.5 Hz, 2H), 4.27 (q, J = 5.8, 5.2 Hz, 2H), 3.93 (dt, J = 11.6, 7.2 Hz, 2H), 1.77 (q, J = 6.4, 4.7 Hz, 2H), 1.30 – 1.22 (m, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 163.46, 144.43, 144.23, 143.81, 134.21, 131.68, 129.90, 127.74, 127.71, 126.88, 126.62, 126.41, 125.22, 124.84, 124.60, 122.71, 122.59, 121.92, 121.83, 121.79, 121.51, 119.93, 116.16, 111.95, 110.83, 110.70, 110.60, 67.77, 43.97, 43.04, 42.46, 27.22, 24.71.
在惰性气氛下, 向Schlenk管中先引入化合物6(0.8 mmol, 500 mg)和碘化锌(0.8 mmol, 255 mg), 加入20 mL超干的THF, 在50 ℃下搅拌20 h后, 冷却后加入正己烷(20 mL), 得到白色固体, 用正己烷(3×30 mL)进行洗涤, 后干燥过夜得到化合物NHC-PDBI-ZnI2(Zn含量: 7.73%(质量分数)).
在惰性气氛下, 向Schlenk管中先引入化合物6(0.8 mmol, 500 mg)和溴化锌(0.8 mmol, 180 mg), 加入20 mL超干的THF, 在50 ℃下搅拌20 h后, 冷却后加入正己烷(20 mL), 得到灰白色固体, 用正己烷(3×30 mL)进行洗涤, 后干燥过夜得到化合物NHC-PDBI-ZnBr2(Zn含量: 7.81%(质量分数)).
在惰性气氛下, 向Schlenk管中先引入化合物6(0.8 mmol, 500 mg)和氯化锌(0.8 mmol, 90 mg), 加入20 mL超干的THF, 在50 ℃下搅拌20 h后, 冷却后加入正己烷(20 mL), 得到白色固体, 用正己烷(3×30 mL)进行洗涤, 后干燥过夜得到化合物NHC-PDBI-ZnCl2(Zn含量: 7.69%(质量分数)).
在惰性气氛下, 向Schlenk管中先加入化合物5b(2.2 mmol, 2 g)和叔丁醇钾(4.5 mmol, 500 mg), 加入50 mL超干THF后, 在40 ℃下搅拌10 h. 冷却至室温后, 加去离子水(50 mL), 用二氯甲烷(3×30 mL)萃取, 用无水碳酸钠干燥后通过真空旋转蒸发浓缩至饱和溶液, 向其中加入正己烷(30 mL), 得到黄色固体, 用正己烷(3×30 mL)进行洗涤, 后干燥过夜得到游离卡宾化合物7(NHC-5b). 由于对应于咪唑环的酸性NCHN信号的消失, 通过1H NMR分析清楚地证实了游离卡宾的形成.
1H NMR (400 MHz, DMSO-d6) δ: 8.40 – 8.27 (m, 4H), 8.22 – 8.10 (m, 10H), 8.01 (q, J = 9.2, 8.8 Hz, 8H), 7.95 (t, J = 3.6 Hz, 2H), 7.76 – 7.64 (m, 4H), 7.42 (d, J = 8.8 Hz, 1H), 7.15 (dd, J = 12.5, 7.3 Hz, 2H), 4.41 (dh, J = 19.1, 5.5, 4.9 Hz, 3H), 4.26 (dt, J = 20.0, 5.6 Hz, 2H), 4.18 – 4.02 (m, 2H), 1.29 – 1.12 (m, 3H), 0.84 (d, J = 7.0 Hz, 1H).
13C NMR (101 MHz, DMSO-d6) δ: 164.04, 153.01, 133.15, 131.68, 131.52, 128.41, 127.81, 127.72, 126.84, 126.57, 126.48, 126.35, 125.66, 125.46, 125.40, 125.21, 124.94, 124.88, 124.80, 124.70, 124.58, 121.50, 121.33, 117.45, 111.04, 68.55, 42.31.
在惰性气氛下, 向Schlenk管中先引入化合物7(0.6 mmol, 500 mg)和溴化锌(0.6 mmol, 135 mg), 加入20 mL超干的THF, 在50 ℃下搅拌20 h后, 冷却后加入正己烷(20 mL), 得到白色固体, 用正己烷(3×30 mL)进行洗涤, 后干燥过夜得到化合物NHC-5b-ZnBr2(Zn含量: 5.75%(质量分数)).
1.4 PO和CO2催化合成环状碳酸酯的反应评价在典型的运行中, PO和CO2的环加成反应在配备有磁力搅拌棒的80 mL高压不锈钢反应器中进行, 依次加入催化剂(0.03 mmol, 0.075%(摩尔分数))和PO(40 mmol), 再充入0.1 MPa的CO2置换3次以排尽反应釜中的空气, 后将CO2引入反应器中并将压力调节至3.0 MPa. 将高压釜在设定的温度下加热到指定的时间段. 反应完成后, 将反应器在冰水浴中冷却至0 ℃, 并释放剩余的CO2. 产物用乙酸乙酯稀释并通过GC分析, 催化剂通过在真空下蒸馏而与产物分离, 直接重复用于下一次运行.
2 结果和讨论 2.1 催化剂的表征由于所合成的氮杂卡宾锌配合物催化剂的结构相似, 我们通过XPS、FT-IR和13C CP-MAS NMR表征手段对催化剂PDBI和NHC-PDBI-ZnBr2的结构进行了分析. 首先, 采用XPS研究了PDBI、NHC-PDBI-ZnBr2、3b、NHC-PDBI-ZnBr2、5b 和NHC-5b-ZnBr2的电子价态和表面元素组成(图2、图3和图4). 从图2(a)可以看出, PDBI表面只存在C、O、N和Br元素, 而在NHC-PDBI-ZnBr2上可以额外观察到Zn元素, 与目标合成催化剂的化学成分一致. 图2(b)−(d)显示了C 1s、N 1s和Br 3d的高分辨率光谱. C 1s光谱(图2(b))分别显示284.8(芳香烃C=C)、285.8(亚甲基碳―CH2―)、286.3(烷基C―O)、286.9(咪唑环C―N)和289.1 eV(咪唑环N―C=N)5个峰. 将Zn 2+引入PDBI中之后, 在283.6 eV处观察到C 1s的新结合能峰, 表明了形成了C―Zn键[30]. N 1s光谱(图2(c))分别对应咪唑环2种不同环境的氮, 其中PDBI的N 1s结合能为401.8 和400.1 eV;NHC-PDBI-ZnBr2的N1s结合能为401.7 和400.0 eV. 在PDBI中引入Zn2+后, 观察到其咪唑环中带正电荷的氮(401.7 eV)的峰显著降低, 说明一些咪唑环与Zn2+转化为氮杂环卡宾配合物[30]. 同时, Br 3d光谱(图2(d))分别显示Br 3d3/2和3d5/2两个能级, 证实了Br−的存在, 有利于对PO的亲核攻击. 对于PDBI, 其Br 3d3/2和3d5/2能级分别出现在68.5和67.5 eV处, 而NHC-PDBI-ZnBr2, 其Br 3d3/2和3d5/2能级分别出现在69.8 和68.8 eV处, 说明在PDBI中引入Zn2+后Br元素的结合能发生了偏移, 这可能归因于在NHC-PDBI-ZnBr2中形成Zn―Br键[30]. 最后, NHC-PDBI-Zn2+的Zn 2p光谱(图2(e)), 在1045.3和1022.4 eV处分别出现对应Zn 2p1/2和2p3/2的结合能, 表明锌以+2价态存在. 以上分析结果表明PBDI和NHC-PDBI-ZnBr2的成功合成. 此外, 从3b、NHC-3b-ZnBr2、5b 和NHC-5b-ZnBr2的XPS谱图观察到类似的结构变化, 进一步表明了氮杂卡宾锌配合物的成功合成.

|
图 2 PBDI和NHC-PBDI-ZnBr2的XPS光谱(a)全谱;(b) C 1s;(c) N 1s;(d) Br 3d和(e) Zn 2p Fig.2 (a) Full spectra, (b) C 1s, (c) N 1s, (d) Br 3d and (e) Zn 2p XPS spectra of PBDI and NHC-PBDI-ZnBr2 |

|
图 3 3b和NHC-3b-ZnBr2的XPS光谱(a)全谱;(b) C 1s;(c) N 1s;(d) Br 3d和(e) Zn 2p Fig.3 (a) Full spectra, (b) C 1s, (c) N 1s, (d) Br 3d and (e) Zn 2p XPS spectra of 3b and NHC-3b-ZnBr2 |

|
图 4 5b和NHC-5b-ZnBr2的XPS光谱(a)全谱;(b) C 1s;(c) N 1s;(d) Br 3d和(e) Zn 2p Fig.4 (a) Full spectra, (b) C 1s, (c) N 1s, (d) Br 3d and (e) Zn 2p XPS spectra of 5b and NHC-5b-ZnBr2 |
图5(a)为PDBI、NHC-PDBI和NHC-PDBI-ZnBr2的FT-IR谱图, 图中出现以下特征FT-IR伸缩峰: (i)在1625 和1358 cm−1处的谱带分别归因于苯并咪唑环的C=N键和C―N键的吸收峰;(ii)在1596 和1557 cm−1处的谱带分别归因于苯环和咪唑环间的共轭振动峰以及苯并咪唑环内的振动峰[31]. NHC-PDBI在1671 cm−1处出现一个强烈的吸收峰, 可归因为苯并咪唑环(NCN)的特征性振动峰. 引入Zn2+后, 该吸收峰红移了9 cm−1, 说明形成了卡宾位C―Zn共价键[31].

|
图 5 NHC-PBDI-ZnBr2、NHC-PBDI和PBDI的 (a)红外光谱和(b)固态13C CP-MAS NMR光谱 Fig.5 Spectra of NHC-PBDI-ZnBr2, NHC-PBDI and PBDI (a) Infrared spectra, (b) Solid state 13C CP-MAS NMR spectra |
固态13C CP-MAS NMR谱(图5(b))在δ值约153和142 处显示两个共振峰, 对应于PBDI苯并咪唑环上的芳族碳, 在δ值约25、50和68处的脂族碳峰归因于两苯并咪唑环之间连接的亚甲基碳和芘与咪唑环之间连接的亚甲基碳[32]. 对比发现在形成游离态卡宾化合物后出现特征性苯并咪唑(NCN)碳峰δ值约在162处. 引入Zn2+后, 在δ值大约171处观察到NCN碳的弱峰, 表明Zn 2+接枝在NHC-PDBI-ZnBr2中部分咪唑环的C2碳上[32].
2.2 催化剂性能评价通过环氧丙烷(PO)和CO2的环加成反应测试了4种不同类型芘标记的NHC前驱体化合物的活性(表1). 在不加催化剂的情况下, 反应几乎不发生(Entry 1). 随着催化剂的加入, 开始检测到一定产物, 其中5a表现出明显优于其他催化剂的催化性能, 可能归因于两个方面, 一方面, 该催化剂比3a和3b有更多的咪唑环作为活性中心, 另一方面, 比5b有更小的空间位阻, 使CO2更容易接近活性中心. 为此, 我们选用5a(PDBI)作为锌基接枝的化合物载体作进一步研究.
| 表 1 不同NHC前驱体化合物对PO和CO2环加成反应的影响a Table 1 Effects of different NHC precursor compounds on PO and CO2 Cycloaddition a |
然后, 考察了卤化锌和NHC前驱体化合物组成的二元催化剂体系以及卤化锌接枝的NHC化合物一元催化体系对于PO和CO2环加成反应的催化活性的影响(表2). 当仅用ZnI2/ZnBr2作为催化剂时, 反应几乎不发生(Entry 1−2), 当仅添加化合物3b/5b作为催化剂时, 反应基本没有活性(Entry 3−4), 添加化合物5a(PBDI)时得到2%的低碳酸丙烯酯收率(Entry 5). 然而, 当5a/3a/5b和ZnX2组成二元催化剂时, PC收率显著提高(Entry 6−10), 表明二者对反应有协同作用. 卤化锌的亲核性以I−>Br−>Cl−的顺序显著影响催化活性, 这与文献报道一致[29]. 令人满意的是, 一元催化剂体系NHC-3b-ZnBr2、NHC-3b-ZnBr2和NHC-PDBI-ZnX2 (X=Cl−、Br−、I−)表现出了较相应二元催化剂体系更优异的催化活性(Entry 6−15), 可能归因于咪唑环的氮杂卡宾位能够吸附活化CO2, 加速PO和CO2的环加成反应. 同时, 观察到催化活性NHC-PDBI-ZnI2 > NHC-PDBI-ZnBr2 > NHC-PDBI-ZnCl2, 这与前面提到的阴离子的亲核顺序相对应. 当NHC-PDBI和ZnI2、NHC-5b和ZnBr2分别物理混合的时, 获得57%和88%的收率(Entry 16−17), 较单组份的NHC-PDBI-ZnI2 和NHC-5b-ZnBr2的催化活性差(Entry 15, 12), 推测可能是由于活性位点之间的相对隔离, 自由接近亲核部分的概率降低, 而由路易斯酸性锌位点和亲核活性位点在同一分子内的催化剂, 更容易攻击合适的位置, 提高反应速率.
| 表 2 不同的锌基NHC化合物对PO和CO2环加成反应的影响a Table 2 Effects of different zinc-based NHC compounds on cycloaddition of PO and CO2a |
以筛选出的最佳催化活性的单组份NHC-PDBI-ZnI2化合物作为PO和CO2环加成反应的催化剂, 系统考察了反应条件如温度、CO2压力、时间和催化剂量等对PC产率的影响. 图6(a)所示, 反应温度对PC产率有显著影响. 催化活性随着温度从80升高至120 °C几乎线性增加, 这归因于在高温下催化剂中的活性物种与底物之间更有效地碰撞[33], 继续升高温度到130 ℃, PC产率从97%缓慢增加到99%. 因此, 选择120 ℃作为优化温度. 反应压力对PC产率也有显著影响(图6(b)). 在0.5~3 MPa范围内, 随着CO2压力的增加, PC产率迅速增加. 当CO2压力高于3 MPa时, PC产率开始下降, 但PC的选择性没有明显变化. 文献中也观察到了压力对催化活性的类似影响[34]. 这可能与催化体系中富PO液相和富CO2气相的相行为有关, 催化反应主要发生在分散于液相中催化剂上,初始CO2压力的增加有助于增加液相中CO2的扩散浓度, 从而有助于促进PO和CO2环加成反应的进行. 然而, 当CO2压力超过3 MPa时, 较高的压力使更多的PO从液相进入气相, 导致催化剂附近的液相PO浓度分布减少, 从而减缓了反应的进行[33−34]. 因此3 MPa是PC合成反应的最佳反应压力条件. 图6(c)为PC产率随着反应时间的变化曲线, 随着反应时间延长, PC产率迅速增加, 2 h后可以达到97%的优异值, 继续延长时间至2.5 h, PC产率缓慢增加至99%. 在反应1 h后移除催化剂, 让滤液继续进行反应, 发现1 h后PC产率缓慢增加到76%, 说明了催化剂相比于其他均相催化剂, 不易溶解于反应体系中, 具有类多相的性质. 在图6(d)为催化剂量对PC产率的影响, 在催化剂与PO百分比在0.025%~0.12%(Mole fraction)的范围内, 随着催化剂量百分比从0.02%增加到0.075%(Mole fraction), PC产率快速增加到97%左右, 随后随着催化剂量百分比增加到0.09% (Mole fraction), PC产率缓慢增加到98%. 催化剂量百分比进一步增加到0.12%(Mole fraction)引起PC产率轻微降低, 推测可能是由于传质阻力增大造成[33]. 因此, 选择0.075% (Mole fraction)作为最合适的催化剂浓度. 最后, 以120 °C, 3 MPa, 2 h, 0.075% (Mole fraction)作为催化剂环加成反应的最佳条件.

|
图 6 反应因素对PC收率和选择性的影响(a)催化剂用量;(b) 反应温度;(c) CO2压力;(d)反应时间 Fig.6 The effect of reaction factors on the yield and selectivity of propylene carbonate (a) catalyst amount; (b) reaction temperature; (c) CO2 pressure; (d) reaction time |
为了考察上述最佳催化剂的普适性, 拓展了其他多种PO底物与CO2环加成反应. 由表3可知, 在相对优化条件下, NHC-PDBI-ZnI2催化剂能够高效催化多种PO与CO2反应生成相应的环状碳酸酯, 可以高选择性和收率转化各种末端PO. 值得一提的是, 环氧己烷和氧化苯乙烯的反应活性略低(Entry 5和Entry 6), 这是由于大的空间位阻阻碍了阴离子对于环氧环的亲核攻击. 由于双环产生的阻碍作用更大, 环氧环己烷相应的环状碳酸酯产率更(Entry 7), 但是, 通过延长反应时间仍然可以获得令人满意的碳酸环己烯酯产率.满意的碳酸环己烯酯产率.
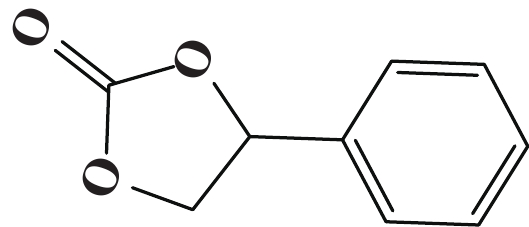
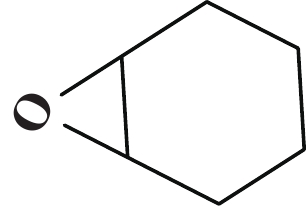
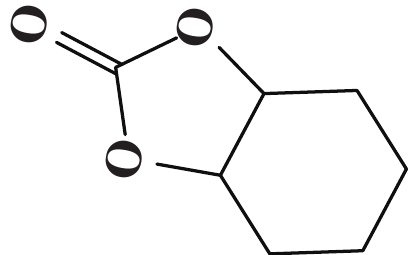
| 表 3 [NHC-PDBI-ZnI2]催化CO2与各种环氧化物的环加成反应a Table 3 Coupling reaction of CO2 with various epoxides catalyzed by [ NHC-PDBI-ZnI2]a |
选取NHC-PDBI-ZnI2在相对优化条件下考察催化剂的稳定性. 在每次循环使用后, 通过减压蒸馏实现了产物和催化剂之间的分离, 并且将所获得的催化剂直接用于下一次重复运行. 如图7(a)所示, 在经过5次循环之后, PC的产率和选择性几乎不变, 表明催化剂具有较优异的稳定性. 采用ICP-AES和FT-IR分别对新鲜和循环后的催化剂进行了结构分析. ICP-AES结果表明, NHC-PDBI-ZnI2催化剂中Zn的质量分数为7.73%, 经过5次循环之后, Zn的质量分数几乎不变(7.71%), 表明催化剂组成稳定. 此外, 由于该催化剂的溶解性不好, 具有类多相催化剂的性质, 进一步采用过滤分离的方式, 考察了催化剂的循环使用性. 如图7(b)所示, 催化剂循环使用3次后PC的收率97%降至20%, 对循环之后的催化剂进行ICP-AES分析发现, Zn的质量分数几乎不变(7.68%), 推测可能是由于催化剂的质量减少引起PC产率降低的. 从FT-IR光谱图(图7(c))发现, 在1700 ~3700 cm−1之间, 每次通过减压蒸馏进行循环后的NHC-PDBI-ZnI2催化剂相比于新鲜的NHC -PDBI-ZnI2催化剂都出现了两个新的C=O键的特征峰, 其中在2278 cm−1处的峰可能是CO2物理吸附的C=O键的特征峰, 在1715 cm−1处的峰可能是氮杂卡宾位吸附CO2的C=O键特征峰[31]. 此外, 从FT-IR光谱图(图7(d))发现, 反应后直接过滤的NHC-PDBI-ZnI2/NHC-5b-ZnI2催化剂相比于新鲜的NHC-PDBI-ZnI2/NHC-5b-ZnI2催化剂也出现了新的C=O键的特征峰, 表明该催化剂的氮杂卡宾位能够吸附CO2.

|
图 7 (a)用NHC-PBDI-ZnI2进行PO和CO2环加成反应的均相循环测试(PO 40 mmol, 催化剂0.075%(摩尔分数), CO2压力3.0 MPa, 温度120 ℃, 时间2 h); (b) 用NHC-PBDI-ZnI2进行PO和CO2环加成反应多相循环测试(PO 40 mmol, 催化剂0.075%(摩尔分数), CO2压力3.0 MPa, 温度120 ℃, 时间2 h); (c) 新鲜的 NHC-PDBI-ZnI2和均质再利用 NHC-PDBI-ZnI2 的红外光谱; (d) 新鲜的 NHC-PDBI-ZnI2和均质再利用 NHC-PDBI-ZnI2 的红外光谱 Fig.7 (a) Homogeneous cycling test of PO and CO2 cycloaddition reaction using NHC-PBDI-ZnI2 (PO 40 mmol, catalyst 0.075%(Mole fraction), CO2 pressure 3.0 MPa, temperature 120 ℃, time 2 h); (b) Heterogeneous cycling test of PO and CO2 cycloaddition reaction using NHC-PBDI-ZnI2 (PO 40 mmol, catalyst 0.075%(Mole fraction), CO2 pressure 3.0 MPa, temperature 120 ℃, time 2 h); (c) FT-IR spectra of Fresh NHC-PDBI-ZnI2 and Homogeneous reused NHC-PDBI-ZnI2; (d) FT-IR spectra of Fresh NHC-PDBI-ZnI2 and Heterogeneous reused NHC-PDBI-ZnI2 |
从FT-IR光谱图可以看出, CO2和PO与NHC-PDBI催化剂反应之后, 在1715 cm−1处出现新的谱带, 其对应于苯并咪唑环新的不对称(C=O)振动, 推测CO2被来自NHC-PDBI-ZnX2的卡宾位吸附活化. 基于上述结果和先前的文献[35], 我们提出了在Zn介导的氮杂卡宾化合物下CO2与PO环加成反应机理(图8). 首先, PO的C―O键被路易斯酸锌中心活化形成锌-环氧化物加合物, 卤素阴离子作为路易斯碱亲核攻击环氧化物的空间位阻较小的β-碳原子, 促进PO的开环[35]. 两者之间的协同作用稳定了中间体含氧阴离子插入CO2. 同时, 来自NHC-PDBI的游离卡宾位吸附活化CO2, 促使形成碳酸烷基酯化合物, 最终分子内环闭合得到环状碳酸酯. 在该催化体系中, NHC-PDBI-ZnX2的氮杂卡宾位和路易斯酸碱位的协同作用促进了反应的有效进行.

|
图 8 NHC-PDBI-ZnX2催化CO2和环氧化物环加成反应的合理机理 Fig.8 Plausible mechanism for the cycloaddition of CO2 and epoxide catalyzed by NHC-PDBI-ZnX2 |
合成了一系列新型芘标记的含溴亲核阴离子的氮杂环卡宾前驱体化合物, 引入卤化锌后, 筛选出优异的氮杂卡宾锌配合物作为单组分多功能催化剂, 在无溶剂和助催化剂的条件下显示出较高的CO2与PO合成环状碳酸酯活性和选择性. 其中NHC-PDBI-ZnI2对CO2与PO的环加成反应表现出最好的催化活性, 并且在其它CO2与PO的环加成反应也表现出了优异的底物适应性. 该催化剂由于形成NHC―Zn强共价键而具有良好的稳定性, 循环5次之后催化活性几乎没有损失. 同时, 基于实验结果和前人的研究提出了一个可能的反应机理, 并推测催化剂的高催化活性原因: (1)路易斯酸性锌物种的极化作用与卤化物对PO的亲核进攻之间的协同作用, 促进了PO的开环; (2)苯并咪唑的氮杂卡宾位有效的活化吸附CO2, 促进其反应成环. NHC-PDBI-ZnX2优异的催化性能和独特的结构有望成为CO2转化多相催化体系的良好候选者.
| [1] |
a. Mikkelsen M, Jørgensen M, Krebs F C. The teraton challenge. A review of fixation and transformation of carbon dioxide[J]. Energy Environ Sci, 2010, 3(1): 43–81.b. Liu Zhen(刘 振), Wu Yu-long(吴玉龙), Nie Ying-fang(聂迎芳), et al. Advances in the synthesis of CO2-Based polycarbonate catalyzed by zinc glutarate(戊二酸锌体系催化合成二氧化碳基聚碳酸酯研究进展)[J]. J Mol Catal(China)(分子催化), 2023, 37(5): 498–511.c. Ye Zhen(叶 朕), Luo Hao-lin(罗皓霖), Jiang Zhi(江 治), et al. Recent advances of photocatalytic CO2 overall reduction(光催化还原二氧化碳全反应的研究进展)[J]. J Mol Catal (China)(分子催化), 2023, 37(2): 174–186.d. Tang Wen-bin(唐文彬), Zhang Zhi-xiang(张志翔), Chi Jia-sheng(池家晟), et al. The state of the art review on the photo-thermal reactor of CO2 reduction(光热催化还原二氧化碳反应器研究进展)[J]. J Mol Catal (China) (分子催化), 2022, 36(5): 499–512.
|
| [2] |
a. Maeda C, Miyazaki Y, Ema T. Recent progress in catalytic conversions of carbon dioxide [J]. Catal Sci Technol, 2014, 4(6): 1482–1497.b. Song Shao-jia(宋少佳), Zhang Xuan(张 璇), Chen Yi-shuang(陈怡爽), et al. Structural-activity relationship of Indium-based catalysts for CO2 oxidative propane dehydrogenation (In基二氧化碳氧化丙烷脱氢催化剂的研究)[J]. J Mol Catal(China)(分子催化), 2022, 36(4): 338–346.
|
| [3] |
Photoelectrochemical conversion of carbon dioxide (CO2) into fuels and value-added products[J]. ACS Energy Lett, 2020, 5(2): 486–519.
DOI:10.1021/acsenergylett.9b02585 |
| [4] |
Phase behaviour study of chalcone in dense CO2[J]. J Supercrit Fluids, 2009, 49(1): 9–15.
DOI:10.1016/j.supflu.2008.11.020 |
| [5] |
Carbon dioxide conversion[J]. ChemNanoMat, 2021, 7(9): 967–968.
DOI:10.1002/cnma.202100240 |
| [6] |
Fully-occupied Keggin type polyoxometalate as solid base for catalyzing CO2 cycloaddition and Knoevenagel condensation[J]. Catal. Sci. Technol, 2016, 6(2): 460–467.
DOI:10.1039/C5CY01038A |
| [7] |
The ability of a zinc pyrrolidine complex to catalyze the synthesis of cyclic carbonates from carbon dioxide and epoxides: A mechanistic theoretical investigation[J]. Dalton T, 2017, 46(28): 9030–9035.
DOI:10.1039/C7DT01642E |
| [8] |
An aminopyridinium ionic liquid: A simple and effective bifunctional organocatalyst for carbonate synthesis from carbon dioxide and epoxides[J]. ChemPlusChem, 2020, 85(7): 1587–1595.
DOI:10.1002/cplu.202000367 |
| [9] |
Chemical CO2 fixation: Cr (III) salen complexes as highly efficient catalysts for the coupling of CO2 and epoxides[J]. J Am Chem Soc, 2001, 123(46): 11498–11499.
DOI:10.1021/ja0164677 |
| [10] |
Cooperative catalyst system for the synthesis of oleochemical cyclic carbonates from CO2 and renewables[J]. Green Chem, 2016, 18(13): 3775–3788.
DOI:10.1039/C6GC00671J |
| [11] |
Bifunctional boron phosphate as an efficient catalyst for epoxide activation to synthesize cyclic carbonates with CO2[J]. Chem-Asian J, 2017, 12(17): 2271–2277.
DOI:10.1002/asia.201700688 |
| [12] |
Zinc-azatrane complexes as efficient catalysts for the conversion of carbon dioxide into cyclic carbonates[J]. ChemCatChem, 2018, 10(4): 843–848.
DOI:10.1002/cctc.201701481 |
| [13] |
Bifunctional porphyrin catalysts for the synthesis of cyclic carbonates from epoxides and CO2: Structural optimization and mechanistic study[J]. J Am Chem Soc, 2014, 136(43): 15270–15279.
DOI:10.1021/ja507665a |
| [14] |
Highly active and robust metalloporphyrin catalysts for the synthesis of cyclic carbonates from a broad range of epoxides and carbon dioxide[J]. Chem-Eur J, 2016, 22(19): 6556–6563.
DOI:10.1002/chem.201600164 |
| [15] |
Cyclic carbonates synthesis by cycloaddition reaction of CO2 with epoxides in the presence of zinc-containing and ionic liquid catalysts[J]. J Iran Chem Soc, 2022, 19(2): 353–379.
DOI:10.1007/s13738-021-02330-9 |
| [16] |
Intramolecularly two-centered cooperation catalysis for the synthesis of cyclic carbonates from CO2 and epoxides[J]. Tetrahedron Lett, 2008, 49(46): 6589–6592.
DOI:10.1016/j.tetlet.2008.09.035 |
| [17] |
A new type of Lewis acid–base bifunctional M (salphen)(M= Zn, Cu and Ni) catalysts for CO2 fixation[J]. ChemCatChem, 2015, 7(10): 1535–1538.
DOI:10.1002/cctc.201500113 |
| [18] |
A recyclable trinuclear Bifunctional catalyst derived from a tetraoxo Bis‐Zn (salphen) metalloligand[J]. Chem-Eur J, 2013, 19(8): 2641–2648.
DOI:10.1002/chem.201204132 |
| [19] |
Multifunctional and sustainable Fe‐iminopyridine complexes for the synthesis of cyclic carbonates[J]. ChemSusChem, 2019, 12(2): 409–415.
DOI:10.1002/cssc.201802563 |
| [20] |
C3-symmetric zinc complexes as sustainable catalysts for transforming carbon dioxide into mono-and multi-cyclic carbonates[J]. Appl Catal B-Environ, 2021, 280: 119395.
DOI:10.1016/j.apcatb.2020.119395 |
| [21] |
N‐heterocyclic carbenes: A new concept in organometallic catalysis[J]. Angew Chem Int Ed, 2002, 41(8): 1290–1309.
DOI:10.1002/1521-3773(20020415)41:8<1290::AID-ANIE1290>3.0.CO;2-Y |
| [22] |
Stabilization of organometallic species achieved by the use of N‐heterocyclic carbene (NHC) ligands[J]. Eur J Inorg Chem, 2005, 2005(10): 1815–1828.
DOI:10.1002/ejic.200500030 |
| [23] |
Organometallic catalysis in aqueous and biological environments: Harnessing the power of metal carbenes[J]. Chem Sci, 2022, 13(22): 6478–6495.
DOI:10.1039/D2SC00721E |
| [24] |
CO2 Hydrogenation catalyzed by a ruthenium protic N-heterocyclic carbene complex[J]. Inorg Chem, 2021, 60(8): 5996–6003.
DOI:10.1021/acs.inorgchem.1c00417 |
| [25] |
Wang Wen-long(汪文龙), Lv Hui(吕 辉), Zhang Guo-dong(张国栋), et al. Autocatalytic click chemistry: Synthesis and characterization of a novel hyper branched "click polymer" based on azacarbene copper complex(自催化点击化学: 新型基于氮杂卡宾铜配合物的超分枝“click 聚合物”的合成与表征)[J]. J Mol Catal(China)(分子催化), 2014, 28(1): 1−6.
|
| [26] |
Zinc imine polyhedral oligomeric silsesquioxane as a quattro‐site catalyst for the synthesis of cyclic carbonates from epoxides and low‐pressure CO2[J]. Chem-Eur J, 2020, 26(60): 13686–13697.
DOI:10.1002/chem.202002996 |
| [27] |
Bifunctional ZIF-78 heterogeneous catalyst with dual Lewis acidic and basic sites for carbon dioxide fixation via cyclic carbonate synthesis[J]. J CO2 Util, 2017, 22: 178–183.
DOI:10.1016/j.jcou.2017.10.005 |
| [28] |
Recent developments on N-heterocyclic carbene supported zinc complexes: Synthesis and use in catalysis[J]. Synthesis, 2018, 50(18): 3662–3670.
DOI:10.1055/s-0037-1610088 |
| [29] |
Periodic mesoporous organosilica with a basic urea‐derived framework for enhanced carbon dioxide capture and conversion under mild conditions[J]. ChemSusChem, 2017, 10(6): 1110–1119.
DOI:10.1002/cssc.201600973 |
| [30] |
CO2 adsorption and conversion into cyclic carbonates over a porous ZnBr2-grafted N-heterocyclic carbene-based aromatic polymer[J]. Appl Catal B-Environ, 2019, 251: 195–205.
DOI:10.1016/j.apcatb.2019.03.076 |
| [31] |
Wang Yan-li (王艳丽), Chen Liu-qing (陈柳青), Liu Xu-guang (刘旭光), et al. Synthesis and spectral properties of 2-(2-Hydroxyphenyl) benzimidazole zinc(2-(2-羟基苯基)苯并咪唑锌的合成及其光谱性能研究)[J]. Funct Mater(功能材料), 2008, 39(6): 971−974.
|
| [32] |
Amino acid/KI as multi-functional synergistic catalysts for cyclic carbonate synthesis from CO2 under mild reaction conditions: A DFT corroborated study[J]. Dalton Trans, 2014, 43(5): 2023–2031.
DOI:10.1039/C3DT52830H |
| [33] |
Polymer nanoparticles grafted zinc-containing ionic liquids: A highly efficient and recyclable catalyst for cooperative cycloaddition of CO2 with epoxides[J]. J CO2 Util, 2018, 28: 96–106.
DOI:10.1016/j.jcou.2018.09.017 |
| [34] |
CO2 cycloaddition reactions catalyzed by an ionic liquid grafted onto a highly cross‐linked polymer matrix[J]. Angew Chem Int Ed, 2007, 46(38): 7255–7258.
DOI:10.1002/anie.200701467 |
| [35] |
Recent advances in the synthesis of cyclic carbonates via CO2 cycloaddition to epoxides[J]. J Environ Chem Eng, 2021, 9(2): 105113.
DOI:10.1016/j.jece.2021.105113 |